5-HMR-2 Chemical Shift
Fortunately for the chemist, all proton resonances do not occur at the same position. The Larmor precession frequency (νo) varies because the actual magnetic field B at the nucleus is always less than the external field Bo. The origin of this effect is the "superconducting" circulation of electrons in the molecule, which occurs in such a way that a local magnetic field Be is created, which opposes Bo (Be is proportional to Bo). Thus B = Bo - Be. We therefore say that the nucleus is shielded from the external magnetic field. The extent of shielding is influenced by many structural features within the molecule, hence the name chemical shift. Since the extent of shielding is proportional to the external magnetic field Bo, we use field independent units for chemical shifts: δ values, whose units are ppm. For an extensive tabulation see Proton shifts.
Spin-spin splitting is not dependent on the external field, so we use energy units for coupling constants: Hz, or cycles per second (in mathematical formulas radians per second are the natural frequency units for both chemical shifts and couplings).
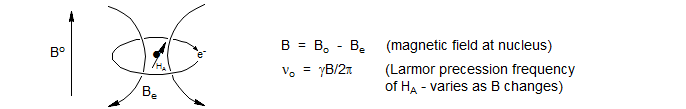
5-HMR-2.1 The Proton Chemical Shift Scale
Experimentally measured proton chemical shifts are referenced to the 1H signal of tetramethylsilane (Me4Si). For NMR studies in aqueous solution, where Me4Si is not sufficiently soluble, the reference signal usually used is DSS (Me3Si-CH2CH2-SO3-Na+, Tiers, J. Org. Chem. 1961, 26, 2097). For aqueous solution of cationic substrates (e.g., amino acids) where there may be interactions between the anionic reference compound and the substrates, an alternative reference standard, DSA (Me3Si-CH2CH2-NH3+ CF3CO2-, Nowick Org. Lett. 2003, 5, 3511) has been suggested.
Proton chemical shifts cover a range of over 30 ppm, but the vast majority appear in the region δ 0-10 ppm, where the origin is the chemical shift of tetramethylsilane.
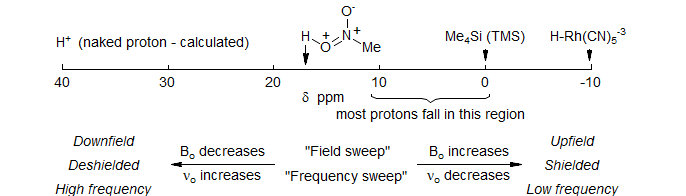
In the original continuous wave (CW) method of measuring NMR spectra, the magnetic field was scanned from left to right, from low to high values. We thus refer to signals on the right as upfield or shielded and signals to the left as downfield or deshielded. Later spectrometers gained the capability of scanning frequency, which then had to decrease from left to right during the scan, hence the "backwards" nature of NMR scales. δ units are defined as follows:
Chemical shifts of all nuclei should be reported using δ values, with frequency and δ increasing from right to left. Many early papers on proton and multinuclear NMR used the opposite convention (not to mention other references) - in particular the τ scale was used in the early days: δ = 10 - τ. IUPAC Recommendations.
The chemical shifts of protons on carbon in organic molecules fall in several distinct regions, depending on the nature of adjacent carbon atoms, and the substituents on those carbons. The scale below should be used only as a rough guideline, since there are many examples that fall outside of the indicated ranges. To a first approximation, protons attached to sp3 and sp carbons appear at 0-5 ppm, whereas those on sp2 carbons appear at 5-10 ppm.

Within these ranges, for a given type of C-H bond (sp3, sp2 or sp) the chemical shift is strongly affected by the presence of electronegative substituents as can be seen in the methyl shifts summarized below, which range from δ -2 for MeLi to δ 4 for MeF.

The 1H chemical shifts of protons attached to heteroatoms (H-X) show a very wide chemical shift range, with no obvious correlation to the electronegativity of X or the acidity of HX. Examples SiH: 1, 2; S-H Se-H Protons.

5-HMR-2.2 Calculation of Proton Chemical Shifts
Parameters for the calculation of proton chemical shifts for many kinds of molecules have been tabulated (see Proton shifts). All of these work in the same way. We establish the base chemical shift for a reference substance (e.g., ethylene for olefins, benzene for substituted aromatic compounds, methane for alkanes) and tabulate Substituent Chemical Shift values (Δδ) for the introduction of substituents into the reference molecules. Thus for a vinyl proton (C=C-H) there will be parameters for the introduction of substituents cis, trans, or gem to the hydrogen we are calculating, and this leads to reasonable estimations for most molecules, as in the example below (parameters from Section 9-HDATA-6.1). Here Δδ 0.15 is the difference between calculated and observed chemical shifts. However, when there are strong resonance or other electronic interactions between substituents (as in the β-aminoenone below, with Δδ 1.70), or strong conformational effects then the predictions made by these calculations will be less accurate. NOTE: the chemical shift increments were determined in weakly interacting solvents like CCl4 and CDCl3. They will work poorly for spectra taken in aromatic solvents like benzene or pyridine (see later section on aromatic solvent shifts).

For aliphatic (sp3) C-H proton chemical shifts we can use the Curphy-Morrison table (Section 9-HDATA-5.1). In this system there are base shifts for CH3 (0.9), CH2 (1.2) and C-H (1.55) protons, and then corrections are applied for all α and β substituents. The corrections for CH3, CH3 and CH protons are slightly different, and no corrections are applied for alkyl groups.

Accuracy of Chemical Shift Calculations
Calculations using simple parameter lists such as in Section 9-HDATA-5.1 and Section 9-HDATA-6.1 will typically give results accurate to within 0.5 ppm, but there are exceptions:
Multiple Substituents: The more parameters you are adding together, and the larger the shift values are, the less accurate the calculation is likely to be. This is especially true for electronegative substituents like O, N and Cl if they are applied several times to the same proton, as in the examples below. This is perfectly reasonable, since electron withdrawal from the C-H group becomes progressively more difficult as the C-H group becomes more electron deficient. Thus, going from methane to methoxymethane causes a +3.3 ppm change in δ, adding a second methoxy, +2.6, and a third methoxy only +0.40 ppm, resulting in progressively larger Δδ errors.

Cyclic Systems: Calculations are usually poorer for cyclic systems, or otherwise conformationally constrained compounds. The base shift for a CH2 group in an alkane is 1.2 ppm, and this would be the calculated value of any methylene group in a cycloalkane. The actual shift for methylenes in cycloalkanes varies by 1.7 ppm, from δ 0.2 for cyclopropane to δ 1.9 for cyclobutane, although if you ignore cyclopropane and cyclobutane, the range is only 0.5 ppm. One of the reasons is that in cyclic compounds conformational mobility is greatly restricted, so that less rotational averaging of various chemical shift anisotropic effects occurs. At low temperatures the axial and equatorial hydrogens of cyclohexane differ by 0.5 ppm, the average shift at room temperature is 1.44, close to the standard value of 1.2. Note especially that the protons on 3-membered rings of all kinds are strongly shifted to lower frequency from the acyclic value.

Even more dramatic chemical shift effects are seen in polycyclic compounds. The Curphy-Morrison calculated values for all of the compounds below would be δ 1.55 (the base value for a methyne group), yet the actual values vary by several ppm. Not surprisingly, cubane and dodecahedrane are especially far from the typical values.

Reproducibility of Proton Chemical Shifts
It is important to understand that the chemical shift of a given proton is not an invariant property of a molecule (like a melting point or boiling point), but will change depending on the molecular environment. The variability is especially large for NH and OH protons (several ppm), but even for CH protons reported shifts vary by a few tenths of a ppm. This is in part due to changes in measurement conditions, but additional variability in chemical shift is present in old NMR data (CW spectra) since spectrometer calibrations and spectrum referencing were not nearly as accurate as they are today. Nevertheless, if conditions are rigorously controlled, very high reproducibility of chemical shifts can be achieved. Databases of precise chemical shifts for many biomolecules have been created which facilitate simultaneous detection by NMR in aqueous solution.
Solvent effects. The aromatic solvents benzene and pyridine cause changes in chemical shifts as large as 0.5 to 0.8 ppm compared to less magnetically active solvents like chloroform or acetone. Since the standard solvent for chemical shift parameters like the Curphy-Morrison ones is CCl4 or CDCl3, expect less accurate calculations for spectra taken in aromatic solvents.
Concentration dependence. Chemical shifts of C-H protons can vary with concentration, especially if intermolecular hydrogen bonding can occur, as for many amines, alcohols and carboxylic acids. The chemical shifts of protons on oxygen (OH) and nitrogen (NH), which are often directly involved in hydrogen bonding are especially strongly dependent (several ppm) on concentration, solvent and temperature. Aromatic molecules can also show significant concentration dependence because of the aromatic solvent effect mentioned above.
Temperature dependence. For molecules that are conformationally flexible, the populations of conformations change with temperature. Since the chemical shifts of various conformations are different, the chemical shifts will vary with temperature (the observed chemical shift is the weighted average of the shifts of the individual conformations). Temperature will also affect the degree of intermolecular hydrogen bonding or other types of aggregation, and this provides an additional source of shift changes.
Paramagnetic impurities (unpaired electrons, transition metals with unpaired spins) can cause very large shifts (tens and hundreds of ppm) as well as large amounts of line broadening. Must avoid these altogether if you want to get high quality NMR spectra.
5-HMR-2.3 Chemical Shift Effects - Electronegativity
Proton shifts move downfield when electronegative substituents are attached to the same or an adjacent carbon (see Curphy-Morrison chemical shift table). Alkyl groups behave as if they were weakly electron withdrawing, although this is probably an anisotropy effect.
The chemical shifts of protons attached to sp2 hybridized carbons also reflect charges within the π system (approximately 10 ppm/unit negative or positive charge).

Even without formal charges, resonance interactions can lead to substantial chemical shift changes due to π polarization.

This is especially useful in the interpretation of the NMR chemical shift of protons in aromatic systems. The protons ortho and para to electron donating and electron withdrawing substituents show distinct upfield and downfield shifts.

5-HMR-2.4 Chemical Shift Effects - Lone Pair Interactions
When lone pairs on nitrogen or oxygen are anti to a C-H bond, the proton is shifted upfield, presumably the result of n --> σ* interaction. There is thus a strong conformational dependence of chemical shifts of protons α to heteroatoms. Whereas for an acyclic tris-amino methane A the Curphy-Morrison chemical shift calculations give a substantial positive error (δcalc - δobs = +2.58), for constrained systems the shifts are highly variable, with the largest upfield shift (-δ) in compound B where there is an enforced anti-arrangement between the lone pair and the C-H bonds (Θ = 180°). This effect is also present in 13C chemical shifts. C-H bonds anti to lone pairs also show Bohlmann bands in the IR spectra, as a result of weakening of the C-H bond by hyperconjugation. Thus compound B shows IR absorption C-H stretch at 2450 cm-1, as well as at 2690-2800 cm-1.

A similar interaction may be responsible for the shift effects noted for the sulfoxides below, where the axial proton anti to the lone pair on sulfur is upfield compared to the stereoisomer.

5-HMR-2.5 Chemical Shift Effects - Steric Compression
When molecular features cause a proton to be forced close to other protons, or to various functional groups, the proton will in general be deshielded (+δ dispersion interactions). Shifts of this type are hard to distinguish from magnetic anisotropy interactions.

These shifts are especially large in highly compressed compounds like the "birdcage" molecules A and C. The inside proton in the "out" alcohol A at δ 4.48 is downfield by 0.96 ppm from the model B. Even more striking are the shifts in the "in" alcohol C, where the proton jammed into the OH group at δ 3.55 is downfield by 2.3 ppm from the model D, and the gem partner at δ 0.88 is actually upfield by 0.5 ppm from its position in D, suggesting a migration of electron density from the sterically compressed inside H to the outside H.

5-HMR-2.6 Chemical Shift Effects - Magnetic Anisotropy
Whereas the local circulation of electrons around a proton is a shielding effect (i.e., to the right in the NMR spectrum, -δ), there can be both shielding and deshielding effects from electron motion in other parts of the molecule. We refer to such interactions as magnetic anisotropy effects, since they are caused by anisotropic electron circulation (i.e., the electron circulation is stronger in some orientations of the molecule in the magnetic field than in others).
The most dramatic examples of anisotropy effects are seen with benzene and other aromatic rings, which cause very large shielding (-δ) effects for protons placed above the ring, and smaller deshielding (+δ) effects for protons to the side of it. These chemical shift effects occur because electron circulation is stronger when the plane of the benzene ring is perpendicular to the magnetic field than when it is parallel to it

The consequence of magnetic anisotropy effects is to provide a stereochemical component to the chemical shift of a nucleus: the chemical shift changes depending on the spacial relationship between a proton and nearby functional groups. Such effects can be valuable for making stereochemical assignments. Some proposed magnetic anisotropy shielding/deshielding cones are shown below:

1. H. C. Brown, A. Suzuki J. Am. Chem. Soc. 1967, 89, 1933. L. A. Paquette, G. Kretschmer, J. Am. Chem. Soc. 1979, 101, 4655.
2. C. D. Poulter et al. J. Am. Chem. Soc. 1972, 94, 2291.
3. The Thiosulfinyl Group Serves as a Stereogenic Center and Shows Diamagnetic Anisotropy Similar to That of the Sulfinyl Group: Tanaka, S.; Sugihara, Y.; Sakamoto, A.; Ishii, A.; Nakayama, J.; J. Am. Chem. Soc., 2003, 125, 9024.
4. Magnetic Anisotropy of the Nitro Group by NMR I. Yamaguchi, Mol. Phys. 1963 , 6, 103
5-HMR-2.7 Chemical Shift Effects - Aromatic Rings
The ring current in Huckel aromatic systems, i.e., those with 4n + 2 π electrons (2, 6, 10, 14, 18 ...) causes downfield shifts in the plane of aromatic ring.

When protons are above or below the plane (or in the middle) of the aromatic ring then large upfield (-δ) shift effects are observed.

When a cyclic conjugated system is planar and antiaromatic, i.e., 4n π electrons (4, 8, 12, 16 ... ) then chemical shift effects are in the opposite direction: downfield over the ring, and upfield in the ring plane. This is seen in the Staley 10 and 12-electron methano annulene cation and anion above, as well as in the Boekelheide 14 and 16-π-electron dihydropyrene below. The normal chemical shift effects are seen in the 10 and 14π-electron systems. In the 12 and 16 π-electron anions the methylene bridge and propyl groups over the ring show very large downfield shifts as a result of the antiaromatic ring current. The paramagnetic ring currents are a consequence of the small HOMO-LUMO separation that is characteristic of 4n π (antiaromatic) systems.

In the Oth [16]-annulene the neutral compound has antiaromatic character. The shifts were measured at low temperature, where conformational averaging has stopped. In the 18π-electron dianion, large aromatic shifts are reported.
Chemical Shift Effects of Phenyl Groups. The presence of phenyl groups in molecules often results in substantial chemical shift perturbations: unusually large diastereotopic effects, odd chemical shifts, and large differences between isomers compared to similar molecules with less magnetically potent substituents. These effects are sometimes predictable and can help with structure assignments. Examples: 1.
The effects of a phenyl substituent are highly dependent on conformation. For example, for styrenes the chemical shift effect of the phenyl is downfield (+δ) when the phenyl is in the plane of the double bond (the usually more stable conformation), but upfield (-δ) when the rotamer with the phenyl group perpendicular is the more stable one, usually as the result of steric interactions:

The large differences in chemical shifts of the butadienes below can be used to assign stereochemistry, based on the upfield effect of the "rotated" benzene ring when it is cis to the other vinyl group (-0.6 ppm).

If steric effects force a phenyl to adopt a face-on conformation (as in the lactone example below) then a cis CH3 group will be shifted upfield compared to a trans group, in this case by 0.4 ppm.

Determination of Enantiomer Ratios and Absolute Configuration with Mosher Esters. Esters of 2-phenyl-2-methoxy-3,3,3-trifluoropropionic acid (Mosher esters, or MTPA esters - α-methoxy-α-trifluoromethylphenylacetic acid) with secondary alcohols show characteristic chemical shift effects in the alcohol portion which can be used to measure enantiomeric purity and assign the absolute configuration of the alcohol. It is necessary to assign key protons, and to make both the R- and S-Mosher ester to arrive at an unambiguous determination (Dale, J. S.; Mosher, H. S. J. Am. Chem. Soc., 1973, 95, 512; Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc., 1991, 113, 4092).
This method works because the principal conformation of MTPA esters is the extended one shown. The anisotropy of the phenyl group then causes upfield shifts of the protons behind the plane of the paper, downfield shifts for those in front. A typical procedure is to do a complete analysis of all assignable protons of the R and S esters, and calculate the difference between the chemical shifts of the two diastereomers. Note that the t-Bu group is upfield in the R,S diastereomer, whereas the Me group is upfield in the R,R isomer.

For a related method using 1-phenyltrifluoroethanol, see Org. Lett. 2003, 5, 1745.

"Chiral Reagents for the Determination of Enantiomeric Excess and Absolute Configuration Using NMR Spectroscopy." Wenzel, T. J.; Wilcox, J. D. Chirality 2003, 15, 256-70. " The Assignment of Absolute Configuration by NMR," Seco, J. M.; Quinoa, R.; Ricardo, R. Chem. Rev. 2004, 104, 17. Parker, D. "NMR Determination of Enantiomeric Purity" Chem. Rev. 1991, 91, 1441
Aromatic Solvent Induced Shifts (ASIS). Polar molecules have substantially different chemical shifts in aromatic solvents (benzene, pyridine, C6F6) than in less magnetically interactive solvents like CCl4, CDCl3, CCl2D2, acetone-d6 and CD3CN. A typical result of going from CDCl3 to benzene is shown in the spectra of butyrophenone below. Other examples: 1, 2, 3, 4, 5, 6, 7, 8, 9. The shifts are large enough that chemical shift calculations can be seriously in error when applied to molecules whose spectra were taken in benzene (P. Laszlo Progr. NMR Spectrosc. 1967, 3, 231).

The origin of these chemical shift effects is believed to be a partial orientation of the solvent by the dipole moment of the solute. For benzene, the shifts can be rationalized on the basis of a weak and transient complexation of the electron-rich π-cloud of the aromatic ring with the positive end of the molecular dipole, such that the protons spend additional time in the shielding (-δ) region above the benzene ring. There is a strong correlation between the dipole moment and the size of the solvent shift, as seen for the series of Sn compounds. With occasional exceptions, the benzene shifts are upfield (-δ).

When 1H NMR spectra are complicated by accidental superposition of coupled protons, as in the spectrum of eugenol in CDCl3 below, then switching to benzene as solvent (or even just adding a few drops of C6D6 to the sample) will often move signals enough that more interpretable (first order) spectra result. In the CDCl3 spectrum of eugenol H2 and H6 are nearly superimposed (WINDNMR simulation gives ΔνAB = 0.4 Hz), leading to a complex ABX pattern of the Solution 2 type. The spectrum in C6D6 removes the near degeneracy of H2 and H6 and converts the strongly coupled NMR spectrum to essentially first order.

In another example, the spectrum below has two near coincidences of protons coupled to each other in CDCl3, which result in the NMR spectrum giving very little information about the structure. In benzene, the spectrum is nearly first order and provides detailed structure information. Note that most (but not all) of the benzene shifts are upfield.

5-HMR-2.8 Chemical Shift Effects - Anisotropy of Double Bonds
C=C Double Bonds. The magnetic anisotropy of C-C double bonds has generally been assumed to be similar to that of aromatic rings, with a deshielding region (+δ) in the plane of double bond. This explains both the downfield (+δ) shifts of vinyl protons, and the larger downfield shifts of the internal (which are affected by the anisotropy of both π systems) versus the terminal protons in conjugated dienes. It also explains the downfield shifts of allylic protons.

The shielding region above and below the plane of the double bond is more controversial. A number of examples show the expected upfield shifts of protons above double bonds.

There is, however, one major exception. In norbornene itself, the proton shifts are in the opposite direction than seen in the 7-substituted norbornenes above (J. Am. Chem. Soc. 1968, 90, 3721). Both the proton assignment and the absence of a -δ region above the double bond are supported by high level ab initio MO chemical shift calculations (J. Am. Chem. Soc. 1998, 120, 11510). Thus the deshielding region above double bonds shown in the figure must be viewed with some skepticism.

For this reason, assignment of stereochemistry in cyclopentanes based on an assumed anisotropy of double bonds, as in the examples below, should be used with caution. Possibly the shifts are the result of C-C single bond anisotropy of the C-vinyl bond.

Anisotropy of Carbonyl Groups. The magnetic anisotropy of C=O has a strongly deshielding (+δ) region in the plane of carbonyl group. This accounts for numerous chemical shift effects in aryl ketones, α,β-unsaturated carbonyl compounds, and conformationally rigid ketones, and is reliable enough to be used for structure assignments.
The effect is seen both when the proton is β to the carbonyl group, as in the enones and acetophenones below, or when there is a γ-relationship.

The tetralone below shows a strongly downfield shifted ortho-proton, to δ 8.0. The ortho-methyl acetophenone, on the other hand shows as smaller downfield shift (δ 7.7), probably due to some rotation of the carbonyl group out of the plane, as well a preference for the conformation shown, with the smaller C=O group cis to the ortho-CH3 group. Acetophenone ortho proton appears at δ 7.9.

In the compounds below, the proton is γ to the carbonyl and close to same plane, leading to quite large downfield (+δ) shifts. The spectrum shows how dramatic the effect can be.




In the stereoisomer A below, one of the aromatic protons is close to the carbonyl, and is shifted downfield by 1.3 ppm, whereas in isomer B the carbonyl is remote, and the chemical shift is normal.

These α,β-unsaturated esters show a shift range of 1.7 ppm resulting from the various β- and γ-carbonyl interactions. In the most upfield shift (δ 6.50 for the E,Z-isomer) there are no close interactions, whereas the most downfield proton (δ 8.20 for the same isomer) has a β-interaction with one carboxylate function, and a γ-interaction with the other:

Amides also show these chemical shift effects. Thus, for the two rotamers of the formamide below, the α-N proton is 0.9 ppm downfield in the isomer with this proton close to the formyl oxygen (Buchi, G.; Gould, S. J.; Naf, F. J. Am. Chem. Soc. 1971, 93, 2492 DOI)

There is some evidence that there is a shielding (-δ) region above the plane of the carbonyl group:

Anisotropy of Nitro groups. The NO2 group may have a a small anisotropic effect similar to that of C=O groups, with a deshielding (+δ) region in the plane of carbonyl group. The ortho protons of nitrobenzenes are strongly downfield, in part due to this interaction. For example the proton Ha between the NO2 and Br groups (the small downfield doublet) has a very similar electronic environment in the two compounds whose spectra are shown below. The upper one has this proton upfield in part because the ortho-methyl group turns the nitro group out of the plane. Of course, turning the nitro group also causes reduced resonance interactions, which causes a shift in the same direction, as seen from the change in the proton ortho to the Me group (Hb).

A similar chemical shift effect in a naphthalene is illustrated below:

5-HMR-2.9 Chemical Shift Effects - Anisotropy of Triple Bonds
Anisotropy of Acetylenes. The magnetic anisotropy of C≡C bonds seems to be well-defined. Both the unusual upfield shift of C≡C-H signals, and the downfield shifts of protons situated next to a triple bond as in the examples below support a strong diamagnetic effect of electron circulation around the triple bond π system.
Anisotropy of Acetylenes. The magnetic anisotropy of C≡C bonds seems to be well-defined. Both the unusual upfield shift of C≡C-H signals, and the downfield shifts of protons situated next to a triple bond as in the examples below support a strong diamagnetic effect of electron circulation around the triple bond π system.

Anisotropy of Nitriles. The cyano group presumably has the same anisotropy as the alkynyl group, as shown by the examples below.

5-HMR-2.10 Chemical Shift Effects - Anisotropy of Single Bonds
C-C Single Bond Anisotropy. Because of the many single bonds in typical organic molecules, each with local anisotropic effects, it has been hard to define single bond chemical shift effects, and even harder to make practical use of them. Nevertheless, useful stereochemical effects have been identified in several situations, loosely based on a magnetic anisotropy of C-C single bonds in which flanking hydrogens are shifted upfield, end-on hydrogens downfield.
Axial and Equatorial Cyclohexane Shifts. In cyclohexane itself, as well as in most substituted and heterocyclic 6-membered rings the axial protons are upfield of the equatorial ones. Unfortunately, there are a few exceptions, and so this chemical shift effect must be used with caution. Below some δe-δa values:

One explanation for this shift effect is based on the anisotropy cones shown in the figure, where the equatorial protons reside in the deshielding (+δ) region of the C-C anisotropy, and the axial in the -δ region. An alternative explanation, or additional contributing effect, is based on the supposition that a C-H bond is a stronger σ donor than a C-C bond, which leads to increased electron density in the axial protons (anti to two C-H bonds), hence -δ. The variation in 1JCH has also been interpreted in these terms.
A more complicated bicyclic ring system shows several shifts that are consistent with the chemical shift effect δeq > δax, and one exception:

Substituent effects on cyclohexanes (Anteunis Tetrahedron Lett. 1975, 687):

Assignment of syn and anti Aldol Adducts. A similar type of single bond anisotropy has been used to rationalize the empirical observation of a systematic variation in the chemical shift of the CHOH proton in syn and anti isomers of aldol products (δsyn > δanti) that can be used to assign configuration, although such assignments should be viewed as less definitive than methods based on coupling constants, because of the usual problem with interpreting small chemical shift differences (Kalaitzakis, D.; Smonou, I.; J. Org. Chem. 2008, 73, 3919-3921). The argument is that in the favored conformation of the hydrogen bonded anti isomer the carbinol proton is in a pseudo-axial orientation subject to similar anisotropy effects as an axial cyclohexane proton, whereas in the syn isomer the proton is pseudo-equatorial.

Cis-Substituent effect in Rigid Rings. Chemical shifts in rigid bicyclic or polycyclic systems can provide some insights into general chemical shift effects, although care must be utilized because there are typically a number of effects operating simultaneously. One example is the tendency for eclipsed or nearly-eclipsed cis-vicinal substituents to cause upfield (-δ) shifts relative to the trans proton (and also relative to the compound with hydrogen replacing the substituent). In the dibenzobicyclo[2.2.2]octadiene system A the cis proton which is eclipsed (or nearly so) with the R substituent is always upfield by 0.5 - 1.1 ppm of the one trans to it, and upfield of the unsubstituted compound as well. The trans protons are all downfield of the model, reflecting inductive effects of the substituents. For the hexachloro bicyclo[2.2.2]heptane B this is also seen, although here the inherent shift difference is not known since the compound with R = H has not been reported.

The upfield shift of cis substituents compared to trans is also seen in a series of succinic anhydrides:

Stereochemical Relations in Cyclopentanes. Because coupling constants are not very reliable for determining stereochemical relationships in 5-membered rings, chemical shift effects such as the one discussed above have been utilized more extensively than in cyclohexanes. It has been observed that in cyclopentanes, γ-butyrolactones (Ollis JCS-PT1 1975, 1480) and tetrahydrofurans the diastereotopic chemical shift effect of a ring CH2 group is consistently larger when flanking substituents X and Y are cis to each other (when the anisotropic effects of the C-X or C-Y bonds are additive) compared to when they are trans (both protons see the effect). More specifically, protons with cis-vicinal substituents are generally shifted to lower δ values (upfield) than those with cis hydrogens. For an example see: Fringenal.

Similarly, the chemical shift of a proton will be a function of the number of cis-alkyl substituents on the ring. To use such chemical shifts it is necessary to have several members of a series for comparison.

Anisotropy of Cyclopropanes. The principal magnetic anisotropy of cyclopropane groups appears to be shielding above the ring and deshielding in the plane of the ring, a ring current effect a little like that of benzene.

Anisotropy of Halogens. Protons positioned near lone-pair bearing atoms such as the halogens generally show downfield shifts, as in the phenanthrene examples below. In dichloro[2.2]paracyclophane the proton adjacent to the chlorine on the other ring, as well as on the side chain are substantially downfield of the other protons. Interpretation of these Δδ values is complicated by the close approach of the X and H atoms, which can cause geometry and orbital distortions that also affect the chemical shifts.


Exercise: Assign all of the protons in the NMR spectrum below.

5-HMR-2.11 Chemical Shift Effects of OH Protons
Hydrogen Bonding Effects. The chemical shifts of OH and NH protons vary over a wide range depending on substrate structure, solvent, temperature and concentration. The shifts are very strongly affected by hydrogen bonding, with large downfield shifts of H-bonded groups compared to free OH or NH groups. Thus OH signals tend to move downfield (+δ) at higher substrate concentration because of increased hydrogen bonding. Both OH and NH signals move downfield in H-bonding solvents like DMSO or acetone.
There is a general tendency for the more acidic OH and NH protons to move further downfield (+δ). This effect is in part a consequence of the stronger H-bonding propensity of acidic protons, and in part an inherent chemical shift effect. Thus carboxylic amides and sulfonamides NH protons are shifted well downfield of related amines, and OH groups of phenols and carboxylic acids are downfield of alcohols.
Recognizing Exchangeable Protons. In many samples NH and OH protons can be recognized from their characteristic chemical shifts or broadened appearance. When this fails, the labile protons can be identified by shaking the sample with a drop of D2O, which results in disappearance of all OH and NH signals. This works best if the solvent is water immiscible and more dense than water (CDCl3, CD2Cl2, CCl4) since the formed DOH is in the drop of water floating at the top of the sample where it is not detected. In water miscible solvents (acetone, DMSO, acetonitrile, pyridine, THF) the OH and NH signals are largely converted to OD and ND, but the DOH formed remains in solution and will be detected in the water region. Examples: 1, 2, 3.
Alcohol OH Protons. In dilute solution of alcohols in non-hydrogen-bonding solvents (CCl4, CDCl3, C6D5) the OH signal generally appears at δ 1-2 At higher concentrations the signal moves downfield (+δ) as a result of increased fraction of H-bonded alcohols, e.g. the OH signal of ethanol comes at δ 1.0 in a 0.5% solution in CCl4, and at δ 5.13 in the pure liquid (from Bovey).

Dynamic Exchange OH. Under ideal conditions OH groups of alcohols can show sharp signals with full coupling to neighboring protons, as in the spectrum of neat ethanol above, and in the spectrum of 1-phenyl-4,4-dimethyl-1-pentyn-3-ol below. To see such coupling pure samples free of acidis or basic impurities are required.Other examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14.

Even long-range coupling can be seen, as in the example below, where the OH proton is a dd, from 3J coupling to the C3 proton (4.3 Hz), and a 4J coupling of 0.7 Hz to one of the diastereotopic C2 protons (presumably a W-type coupling).

More typically, signals for OH protons are subject to rapid (on the NMR time scale) intermolecular exchange processes, which may result in broadening or complete loss of coupling to neighboring protons. In the example below, the ABMX3 signal H1 at δ 3.9 is broadened by partial averaging of coupling to the OH group. Note that the ABMX3 signal (H2) at δ 4.7 is sharp. Other examples 1, 2, 3, 4.

Such exchange can also broaden or average the signals of multiple OH, NH or SH groups in the sample, if more than one is present. Any water present might also exchange with the R-OH protons. The rates of exchange are a complex function of temperature, solvent, concentration and especially the presence of acidic and basic impurities. In CDCl3 the presence of acidic impurities resulting from solvent decomposition often leads to rapid acid catalyzed exchange between OH groups.
Solvents like DMSO and acetone form strong hydrogen bonds to the OH group. This has the effect of slowing down intermolecular proton exchanges, usually leading to discrete OH signals with observable coupling to nearby protons. Note the triplet and doublet for the HOCH2 group in the spectrum below taken in DMSO.

In the remarkable NMR spectrum of the OH region of sucrose below taken in aqueous acetone solvent all of the OH signals and their coupling are resolved (Adams, Lerner J. Am. Chem. Soc. 1992, 114, 4828).

Phenol OH Protons. The OH signals of phenols are generally well downfield of those of alcohols, appearing at δ 5-7 in CDCl3, and δ 9-11 in DMSO. The higher acidity of phenols results in faster exchange rates, so that polyphenolic compounds will usually show only one OH signal.
In DMSO solution, even the exchange between carboxylic acid protons and other OH groups can be slowed enough to allow individual observation, as in the spectrum of 2-hydroxycinnamic acid below.

β-Dicarbonyl Compounds. Especially dramatic shifts are observed for the strongly intramolecular H-bonded enol forms of β-dicarbonyl compounds, ortho-ketophenols and related structures. Examples: 1.

Carboxylic Acids. Most carboxylic acids are strongly hydrogen bonded in non-polar solvents, and the OH protons are correspondingly downfield (+δ) shifted. Acetic acid dimer in Freon solvent (CDClF2/CDF3) at 128 K appears at δ 13.04, and the OH signals of acetic acid hydrogen bonded to a protected adenosine under conditions of slow exchange appear at even lower field (Basilio, E. M.; Limbach, H. H.; Weisz, K. J. Am. Chem. Soc. 2004, 126, 2135). Other Examples: 1

5-HMR-2.12 Chemical Shift Effects of NH Protons
Amine N-H protons: NH2 protons of primary alkyl amines typically appear as a somewhat broadened signal at δ 1-2 in CDCl3. The broadening has several sources: partially averaged coupling to neighboring protons, intermolecular exchange with other NH or OH protons, and partially coalesced coupling to the quadrupolar 14N nucleus (I = 1), which usually has a short T1. In the example below, the CH2 group bonded to amino (δ 2.82) shows little indication of coupling to the NH2 protons, so NH exchange must be rapid on the NMR time scale. Other examples: 1, 2, 3, 4, 5. The amide proton at δ 7.1 is in slow exchange (the vicinal HN-CH2 and CH2-CH2 couplings are accidentally equivalent). It is broadened by residual coupling to 14N, not by exchange, since the N-CH2 signal is a sharp quartet.

Ammonium N-H protons. The N-H signals of ammonium salts are strongly downfield shifted, typically appearing at δ 4-7 in CDCl3 and δ 8-9 in DMSO. If spectra are taken in strongly acidic solvents (e.g. trifluoroacetic acid), where intermolecular exchange is slowed, the signals are sometimes very broad, and can show poorly resolved 1H-14N J coupling (1:1:1 triplet, JHN ≈ 70 Hz). Under these conditions N-H protons in ammonium salts show coupling to neighboring C-H protons.

Exercise: Rationalize the coupling in the partial NMR spectrum below.

Aniline NH Protons. The NH protons of anilines are typically at δ 3.5-4.5 in CDCl3 solution, moving downfield by 1-2 ppm in DMSO solution. ortho-Nitroanilines (ca δ 5-6) and heterocyclic amines such 2-aminopyridines (δ 4.5) have signals downfield of this range. Examples: 1, 2, 3.

Amide NH Protons. Amide NH signals typically appear around δ 7, as in the example of N-acetylethylenediamine above and N-methylpropionamide below. They are generally in slow exchange with other NH and OH signals. Thus, neighboring protons will show coupling to the NH proton, as in the examples, where the CH2 bonded to the amide nitrogen is a quartet and the N-Me group is a doublet. The amide N-H protons are typically broad from poorly resolved coupling to 14N, so the coupling to neighboring protons is usually not resolved, or barely resolved in the NH signal. Examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.

5-HMR-2.13 Chemical Shift Effects of SH, SeH and TeH Protons
Thiol S-H Protons. S-H protons of alkyl thiols typically appear between δ 1.2 and 2.0 in CDCl3. The position is not strongly affected by hydrogen bonding solvents like acetone or DMSO, since SH protons are only weakly hydrogen bonded. Coupling to nearby protons is usually seen, although broadened or fully averaged signals are not uncommon, especially in molecules containing OH protons (or in impure samples). Examples: 1, 2, 3.


Aryl thiol S-H signals are further downfield, typically δ 3.5-4.5, as a result of normal ring-current effects, and the greater electron withdrawing effect of aryl vs.alkyl groups.

Selenol and tellurol protons (SeH and TeH) behave like thiol protons, but appear further upfield -- around δ 0 for SeH and δ -3 to -5 for TeH. Below a comparison of the NMR spectra of benzylselenol and benzylthiol. Note that both the Se-H and S-H protons are coupled to the CH2 group (AX2 pattern) - intermolecular exchange rates are usually much slower for SH and SeH protons than for OH protons. Both selenium and tellurium have NMR-active spin ½ isotopes, so in the Se-H proton we can see the satellites due to coupling between 1H and the 7.5% of the spin 1/2 isotope 77Se.

5-HMR-2.14 Hydrogen Fluoride Species
At very low temperatures in Freon solvents it has been possible to characterize a number of H-bonded HF species (Shenderovich, Tolstoy, Golubev, Smirnov, Denisov, LimbachJACS 2003, 125, 11710). A few examples are shown below.

The shifts and coupling constants are temperature dependent. This is because at lower temperatures the solvent dipole moment increases, and this can distort these soft structures. For example, 1JHF in B goes from 66 Hz at 170 K to 15 Hz at 130 K. Presumably the structure is moving from N---H-F at higher temperature towards the more strongly dipolar structure N-H---F at lower ones. Note that 2JNF is unchanged.
Next Section: Coupling · Previous Section: Integration · Home