5-HMR-5 Vicinal Proton-Proton Coupling 3JHH
The single most useful H-H coupling relationship is that between vicinal protons. The size of 3JH-H is predictable and provides detailed information about the spacial orientation between the two protons. Almost all 3JHH values are positive (a rare exception is the -2 Hz 3JH-H in cis-1,2-difluoroethylene), but their magnitude varies widely (from close to 0 Hz up to 25 Hz) depending on structural and conformational details.
Three-Bond Coupling across Single Bonds. In acyclic systems with small conformational preferences, vicinal couplings are generally in the range 6-8 Hz, with electronegative substituents causing smaller J values (see the Pauling electronegativities E in the graphic). Note in particular the reduced 3J for protons on carbons bearing oxygen substituents (as well as F), which is seen for all types of 3-bond couplings (Review: Bothner-By Adv. Magn. Reson. 1965, 1, 195.)
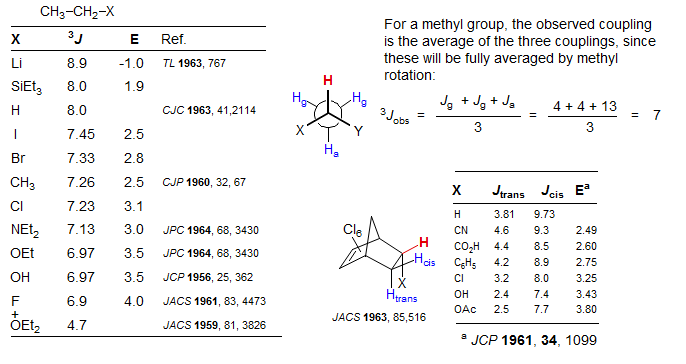
When there are two electronegative substituents the vicinal coupling is reduced further, although adding a third such substituent does not seem to affect the coupling much.
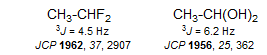
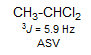
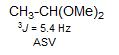
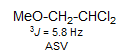
5-HMR-5.1 The Karplus Equation
The Karplus equation (J. Chem. Phys. 1959, 30, 11)) is based on the observation, supported by theoretical considerations, that vicinal H-H couplings will be maximal with protons with 180° and 0° dihedral angles (anti or eclipsed relationship results in optimal orbital overlap) and that coupling will be minimal (near 0) for protons that are 90° from each other. The equation gives us approximate values for 3JHH as a function of dihedral angle between the protons. It should be remembered, however, that this relationship strictly applies only in unstrained hydrocarbon systems, and that electronegative substituents and ring constraints may cause substantial perturbations (in both positive and negative directions) to the values predicted by this equation. Nevertheless, the Karplus curve (together with more complicated variants) is the mainstay of conformational analysis for all ring systems, and has generally proved reliable if care is taken. The constants Jo and K are used to correct for substituent effects in more sophisticated uses of the Karplus equation, different Jo values are also used for the 0 to 90° and the 90 to 180° sections of the curve.
A more sophisticated molecular orbital treatment led to a modified Karplus equation (J. Am. Chem. Soc. 1963, 85, 2870) that does not require different J0 values for the 0-90 vs 90-180° sections. We will use this equation in our discussions here.
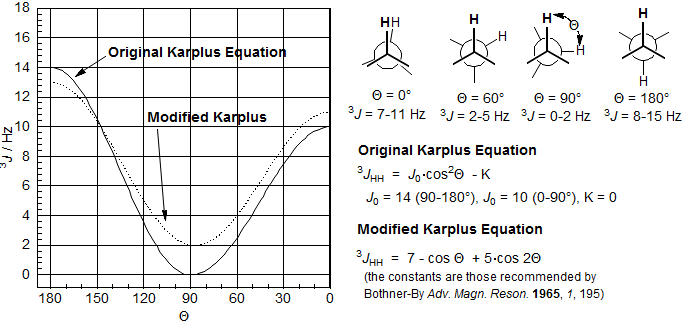
A convenient graphical form of the Karplus relationship is given in Figure 5.5.1 below. Here two curves, separated by 120°, represent the predicted coupling constants for a proton H1 coupled to an adjacent methylene group (Hcis and Htrans), as a function of the dihedral angle.

Figure 5.5.1. Double Karplus Curve for Vicinal coupling in Cycloalkanes.
5-HMR-5.2 Determination of Stereochemistry in Cyclohexanes Using 3JHH
It is often straightforward to establish stereochemical relationships among substituents in cyclohexanes, provided that the spectrum can be analyzed. In chair cyclohexanes, the relationship among vicinal protons is restricted to the narrow regions for Θ1-c = 40-60 on Figure 5.5.1 (i.e. to the left of the H1-eq crossing point at 60°, and to the right of the H1-ax point). These regions correspond to flattening of the cyclohexane, which is energetically easy. The opposite distortion (Θ1-c = 60-85) cannot occur to any significant extent. Jaa is usually much larger (9-12 Hz) than Jee or Jea (each usually 3-4 Hz).
Below is reproduced the 100 MHz NMR signal of the H1 proton of iodocyclohexane at -80°C (from F. R. Jensen, C. H. Bushweller, Beck JACS 1969 91, 344, 3223). Under these conditions the ring inversion is slow on the NMR time scale, and separate signals are seen for the two conformational isomers. The couplings are not always this well resolved, but the axial proton multiplet will almost invariably be much wider than the equatorial one (remember that the separation of the outer two lines of a first order multiplet is the sum of all the coupling constants). At room temperature, the ring inversion will be fast on the NMR time scale, so an average spectrum will be observed. It will look much like that of the axial proton, since the equatorial isomer is the major one. Examples of J in 6-membered carbocycles: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11. 6-Membered heterocycles: 1, 2, 3, 4, 5, 6, 7, 8. See section on sugars and cyclohexenes for more examples.

A more detailed look is provided by octa-deuterochlorocyclohexane, taken at 60 MHz with deuterium decoupling. Here only the ABX system of the protons at C1 and C2 are present. At -76 °C the ring inversion has become slow on the NMR time scale, so distinct signals are seen for the axial and equatorial conformations, with the characteristic small 3Jeq-eq and 3Jax-eq, and the large 3Jax-ax (Hoefner, Lesko, Binsch Org. Magn. Reson. 1978, 11, 179).

The coupling constants in cyclohexane itself were determined by analysis of the AA'BB' pattern of 1,1,2,2,3,3,4,4-octadeuteriocyclohexane at -103 °C (Garbisch, J. Am. Chem. Soc. 1968, 90, 6543). The bottom spectrum (deuterium decoupled) is the experimental one, the top one is a simulation with the parameters listed.

The spectra of iodocyclohexane and cyclohexane itself also illustrate another feature common to many axial and equatorial cyclohexane protons: the chemical shift of the axial proton is usually upfield of the equatorial one, in the case of cyclohexane by 0.5 ppm.
The near identity of the magnitudes of the gem (2JAB = -13.05 Hz) and axial-axial (3JAA' = +13.12 Hz) couplings seen in cyclohexane is a common feature of substituted chair cyclohexanes and half-chair cyclohexenes. Note that the couplings do have opposite signs, although this is not detectable in first-order spectra. In molecules with electronegative substituents (e.g. pyranose sugars, Section 5-HMR-05.2 ) the vic axial-axial couplings are smaller than these, with typical values between 8 and 11 Hz.
In an idealized cyclohexane, Jee and Jae are expected to be nearly identical, since each corresponds to a dihedral angle of 60°. However, cyclohexanes are typically slightly flattened, presumably due to axial-axial repulsions. This moves the dihedral angle for Jee to slightly higher than 60°, hence smaller coupling, and that of Jae to slightly below 60°, resulting in larger coupling (see the shaded areas in Figure 5.5.2). The dihedral angle in cyclohexane itself is 57°, and this leads to the slightly smaller value for Jee (JBB' = 2.96) compared to Jae (JAB' = 3.65). Similar effects are also commonly seen in substituted cyclohexanes which are conformationally homogeneous, especially if there are axial substituents of any size.
If the flattening of a cyclohexane is substantial, Jee can become too small to detect (as is the case for some bicyclo[3.3.1]nonanes and bicyclo[321]octanes with Θ1-t = 90°), and Jae can become substantially larger than the normal values of 3-4 Hz, reaching values of 5 or even 6 Hz. Thus you cannot always rely on getting an exact count of vicinal neighbors to a proton from its multiplicity. Examples: 1, 2, 3.
The near identity of Jee and Jea has the unfortunate consequence that the couplings to an equatorial proton do not provide information about the stereochemistry of neighboring protons (i.e. whether they are axial or equatorial) although they will usually provide a count of the vicinal neighbors.

Figure 5.5.2. Karplus Curve (3J = 7 -cos Θ + 5·cos 2Θ) for vicinal coupling in cycloalkanes. The shaded area represents the typical conformational space of chair cyclohexanes, showing ring flattening.
Exercise: The 1H NMR spectra of two isomers of methyl 2-(N-benzoylamino)cyclohexanecarboxylate are show below. Determine which isomer corresponds to spectrum A and B, and which conformation is the major one for each. Focus on an assignment and complete analysis of the three downfield protons corresponding to the N-H, α-carbomethoxy and α-aminobenzoyl protons.



Exercise: Analyze the NMR spectrum of the mixture of 3,5-diphenylbromocyclohexanes below (assign signals):

Boat Conformations. The twist boat conformation in cyclohexane is ca 5 kcal higher in energy than the chair, so it requires a substantial perturbation to force a chair into a boat. Even cyclohexanes with a tert-butyl groups forced to be axial can adopt modestly distorted chair conformations. As a consequences, twist-boat cyclohexanes are very rare, commonly seen only in bicyclic structures, or in 6-membered rings with multiple heteroatoms (e.g. 1,3-dioxanes) or those containing multiple sp2 carbons where axial repulsions are minimal.

In boat and twist-boat cyclohexanes there are multiple conformations, each of which have available several C-C-C-C dihedral angles. In an idealized twist-boat there are four kinds of hydrogens, with eight dihedral angle relationships (ca 30, 30, 50, 50, 70, 90,150, 170 degrees). In addition, there are six different twist boats possible for a multiply-substituted cyclohexane so stereochemical assignments are very difficult.
Cyclohexenes. The local conformation of the four sp3 carbons in cyclohexenes are very similar to those in cyclohexane, and very similar coupling constants are seen - 3Jax-ax is large (typically 10-12 Hz), whereas 3Jax-eq and 3Jeq-eq are small (typically 2-4 Hz). Examples: 1, 2, 3, 4, 5.

Problem R-84I (C9H14O2). Shown below is the partial NMR spectrum (100 MHz, CS2 solvent, deuterium decoupled) of a deuterated cyclohexene derivative (the CH3 resonances are not shown). Assign signals and determine the stereochemistry (cis or trans). Source: Org. Magn. Res. 1973, 5, #10, Spect #0635

For 3J allylic coupling in cyclohexenes see Sect. 5-HMR-5.7.
Cyclohexanones. The gem coupling α to the carbonyl group is slightly larger in cyclohexanones than in typical cyclohexanes, consistent with the generally observed effect of π-acceptors on 2J. The vicinal coupling constants α and β to the carbonyl group are modified by the flattening effect of the sp2 carbon, specifically 3Jax-eq is larger (typically 4-6 Hz) than 3Jeq-eq (typically 2-3 Hz). 3Jax-ax is normal 12-13 Hz. Examples: 1, 2, 3.

Stereochemistry in 6-Membered Heterocycles
All of the considerations that apply to cyclohexanes also apply to tetrahydropyrans, piperidines, thianes and many other 6-membered heterocycles. Most are overwhelmingly in the chair form, although twist-boat conformation are energetically more accessible than for cyclohexanes because of the smaller number of axial substituents (lone pairs are less sterically demanding than H or other substituents). The spin systems are usually simpler, allowing easier interpretation of the multiplets.Examples of 3J in 6-membered heterocycles: 1, 2, 3, 4, 5, 6, 7, 8.
Sugars. The pyranose (6-membered ring) forms of pentose and hexose sugars provide many examples where vicinal proton coupling constants allow complete assignment of stereochemistry. Analysis of sugar NMR spectra always begin with the specific assignment of one or more of the protons, either from chemical shift information or the number of couplings. In the example below, the best place to start is H1, which can be recognized both from its chemical shift (at δ 6.6), as well as from the observation that it will be the only proton in the molecule coupled to just one other proton. For other examples of sugars and sugar-like molecules see: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.
Exercise: Examine the 300 MHz spectrum of glucose pentaacetate reproduced below. Assume you don't know the stereochemistry and use the spectrum to assign it at each of the carbons. Click spectrum for answer.

Exercise (R-05I). The multiplet below corresponds to two of the protons of compound R-05I (C13H16O2). Analyze and assign the multiplet, report couplings and δ values, and determine the stereochemistry and conformation of the compound (add appropriate substituents and protons to the structure on the right). Briefly explain your reasoning. Click spectrum for answer

Cycohexanes and 6-membered heterocycles with enforced axial-axial repulsions are usually flattened to some extent, which can lead to small or undetectable eq-eq couplings. A nice example of ring flattening and the resultant "missing" eq-eq 3J is shown for an isomer of the alkaloid cocaine shown below. The NMR spectrum fragment shows the signal for the proton H3, which is just a doublet. The one coupling observed is 3J3e-4a, and it is, as expected, unusually large (6.2 Hz) for an ax-eq 3J. The other two couplings (both eq-eq) to H-2e and H-4e are too small to detect at this resolution.

Exercise: Below is shown the multiplet for H3 of another isomer of Cocaine. Interpret the multiplet and identify the structure

Exercise: Assign the seven protons indicated and determine all coupling constants in the spectrum of the furyl-substituted oxathiane ring. All multiplets are basically first order, except for a little leaning. The multiplets present are listed to help out with the analysis. Click spectrum for answer

5-HMR-5.3 Determination of Stereochemistry in Cyclopentanes Using 3JHH
The conformational analysis of substituted cyclopentanes is much more complicated than that of cyclohexanes. The energy differences between various envelope and twist conformations in five-membered rings are generally small, and there are as many as ten different envelope and ten different twist conformations, and each conformation has multiple dihedral angle relationships. Several of the 20 possible conformations may be populated in an individual structure. Thus the vicinal couplings in 5-membered rings are highly variable. For cyclopentanes in envelope conformations Jcis > Jtrans in the flat part of the envelope, whereas in twist conformations the tendency is for Jtrans > Jcis. In general, no firm assignments of stereochemistry can be made using the size of couplings alone unless a specific substitution pattern or heterocyclic system has been carefully investigated, or if substitution patterns allow prediction of the conformation. The shaded parts of the double Karplus curve below illustrates the coupling constant ranges typically available in cyclopentanes and saturated 5-membered heterocycles. Examples - 5-ring: 1, 2, 3, 4, 5, 6 7, 8, 9, 10.

Inspection of the double Karplus curves indicates a significant difference between the typical behavior of adjacent CH2 groups in cyclohexanes and cyclopentanes. In a chair cyclohexane only one of the four vicinal couplings can be large (trans ax-ax, > 7 Hz), whereas in a cyclopentane it is common for 2 or even 3 of the 3J couplings to be large. When there is a small (<4 Hz) 3J coupling in a cyclopentane or saturated 5-ring heterocycle, the protons are often in a trans relationship. That is not to say that pseudo-equatorial protons do not sometimes have small coupling to both protons of an adjacent CH2 in cyclopentanes, but it is less common than in cyclohexanes.

The contrast between 6- and 5-membered tings is illustrated by the examples below, where H1 and H2 in the 6-membered heterocycle show only one large 3J coupling (red, ax-ax) and three small couplings (blue), whereas the 5-membered heterocycle shows three large couplings (red) and one small (blue). The other large coupling is the one between H1 and H2 (2J).

In most cyclopentanes, the C-C-C-C dihedral angles are significantly smaller than the 60° found in cyclohexanes. Cis protons will tend to have H-C-C-H dihedral angles close to 0°, and trans near 120°. The cis couplings (8-10 Hz) are usually larger than trans (2-9 Hz). However the Karplus curves for cyclopentane have a region where the cis and trans lines cross (Figure above, at ca 20° dihedral angle), so there are many cases where Jtrans > Jcis. There are also cases where cis and trans couplings are identical, as on the compound below, where the allylic proton is a quartet of doublets, arising from accidental equivalence of three vicinal couplings.

This signal provides a word of warning about jumping to conclusions during the interpretation of coupling patterns - it would be reasonable to assume that the proton responsible for this multiplet is definitely coupled to a methyl group and one other proton by a small coupling.
In the above compound the cis and trans coupling in the five membered ring are nearly identical. This arises from a small degree of puckering with the substituent in the pseudo-equatorial position to avoid 1,3-diaxial interactions with the methyl group.
The NMR spectrum of the ring protons of an acylated Evans oxazolidine chiral auxiliary show very different coupling - there must be a small pucker at the C-O or C-N carbon, with the substituent pseudo-axial, leading to 3Jcis of 8.5 Hz and a 3Jtrans of 2.9 Hz (these were obtained from a simulation with WINDNMR, first order analysis as an AMX system leads to 8.7 and 3.4 Hz, showing the typical partial averaging of the two couplings in a strongly coupled spin system).

Stereochemical relations among vicinal protons in 5-membered rings cannot be reliably determined by simply measuring coupling constants, except in cases where the substitution pattern of the specific ring system has been carefully investigated. For example, in the benzodihydrofurans below, changing the size of the substituent R causes a reversal in the size of Jcis and Jtrans. If the ring puckering is strong enough, then Jtrans > Jcis.

In bicyclo[2.2.1]heptanes the endo-endo and exo-exo 3J (cis, Θ ≈ 0) are always greater than endo-exo (trans, Θ ≈ 120) couplings.
Coupling constants within furanose sugars often do not define the stereochemistry (this is in sharp contrast to pyranose forms), although if the stereochemistry is known, some insight into conformation can be obtained. The opaque nature of the coupling is illustrated for Thymidine, where there are two cis (AE and AD) and and three trans couplings (AE, AD, CD) within the furanose ring. All four couplings are nearly the same size (5-7 Hz)

A somewhat unusual pyrrolidine case is cis-4-hydroxyproline, where the NMR evidence indicates that both substituents (the OH and the CO2H) are in pseudo-axial positions, so that there are an unusual number of small couplings among the ring protons. As a result, the cis 3J (3.8 to 10.3 Hz) are all larger than the trans 3J (1.6 to 3,7 Hz). A 4J W-type coupling between H3 and H6 is as large as one of the trans 3J couplings (that between H6 and H4). A summary of the observed couplings (from a WINDNMR simulation) is shown below the spectrum.

Although such situations are not commonly seen, very small 3J do occur, particularly in polycyclic systems where dihedral angles near 90° are rigidly enforced, and other factors which reduce 3J coupling (such as strained rings) are present. An example is provided by the partial NMR spectrum of 3-aza-bicyclo[3.1.0]hexane-2-carboxylic acid below, where both protons cis to the fused cyclopropane have no detectable coupling to the trans vicinal cyclopropyl proton.

Exercise: Determine coupling constants, assign them, and propose a conformation for the compound below. Click spectrum for answer.

Exercise: This is a mixture of cis and trans isomers of the enol ether shown. Assign all of the proton signals, and extract coupling constants. First order analysis is appropriate. Which is the major isomer? Click spectrum for answer.

5-HMR-5.4 Determination of Stereochemistry in Cyclobutanes Using 3JHH
Cyclobutanes are even flatter than cyclopentanes, so that cis couplings are almost always larger (6-9 Hz) than trans (2-8). However, if structural features which promote strong puckering of the ring such as a trans ring fusion, large or electronegative substituents are present, then trans couplings can become larger than cis, as shown for cis-1,3-dibromocyclobutane and cyclobutanol below.



5-HMR-5.5 Determination of Stereochemistry in Cyclopropanes Using 3JHH
Dihedral angles in cyclopropanes are rigidly fixed by the geometry of the ring system. Thus Jcis (7-13 Hz) is always larger than Jtrans (2-7 Hz), and this can be reliably used for structure assignment. Examples: 1, 2, 3, 4, 5, 6.

The same relationship holds for the 3-membered ring heterocycles, although the range of observed couplings is wider. Examples: 1, 2, 3, 4, 5, 6.


Exercise: The two partial 1H NMR spectra below correspond to the E and Z isomers of ethyl cinnamate epoxide. Which is which? Explain the appearance of the multiplet at δ 4.25/3.95.


Summary: On the double Karplus curve below are indicated the dihedral angles and hence the cis and trans 3-bond couplings that can be observed for various rings. Chair cyclohexanes are conformationally well-defined, with a relatively small range of 3J couplings possible (Jeq-eq and Jeq-ax typically 3-4 Hz, and Jax-ax typically 8-13 Hz). With 5 and 4 membered rings a wider range of couplings are seen depending on the extent and type of puckering present. Cis couplings will typically be larger than trans couplings. Unfortunately for both cyclopentanes and (less commonly) cyclobutanes, Jtrans can occasionally be larger than Jcis for pseudoaxial protons, if the conformation places the dihedral angle to the left of the crossing point at ca 20°. For such systems both Jtrans and Jcis will be relatively large (8-10 Hz). Cyclopropanes are rigid, and Jcis (eclipsed, Θ = 0°) is always greater than Jtrans (Θ = 120°). With this in mind, the appearance of only well defined large (ca 10 Hz) and small (ca 3 Hz) in a CH coupled vicinally to one or more CH2 groups is quite characteristic of cyclohexanes. Cyclopentanes and cyclobutanes, on the other hand, tend to more frequently have intermediate size couplings (5-9 Hz), and often nearly equal and large coupling to cis and trans vicinal neighbors.

The graphic below summarizes vicinal coupling in sample 6, 5, and 4 membered rings, illustrating the contrast between 6-rings, where there is always only one large coupling (red) between adjacent CH2 groups (trans diaxial), whereas in 5-membered rings (especially those with one or more sp3 atoms) and 4-membered rings, there is often only one small coupling (blue), which is usually between trans-protons.




5-HMR-5.6 Acyclic Stereochemistry using 3JHH
Syn-Anti Stereochemistry of Aldol Adducts. The use of 3J for conformational analysis in acyclic systems can be more difficult than within rings because of the larger number of conformations typically possible (Review: "Determination of Relative Configuration in Organic Compounds by NMR Spectroscopy and Computational Methods" Bifulco, G.; Dambruoso , P.; Gomez-Paloma, L.; Riccio, R. Chem. Rev., 2007, 107, 3744 DOI: 10.1021/cr030733c). Basically, a single coupling constant cannot usually distinguish which of two diastereomers might be present since there are 3 possible staggered conformations for each diastereomer, two of which will typically have very similar predicted coupling constants for a pair of vicinal protons. Assignments become possible only when one can make some reliable predictions on which conformation predominates. One such situation is encountered with the diastereomeric products of an ethyl ketone aldol reaction, as shown below:

In non-polar media, a hydrogen bond between OH and the carbonyl group is expected. Since in the syn isomer both hydrogen bonded conformations have a gauche relationship between HA and HB, we expect a smaller 3J for the syn isomer than for the anti, where one of the H-bonded conformations has an anti relationship between HA and HB (Stiles-House rule: Stiles J. Am. Chem. Soc. 1964, 86, 3337; House J. Am. Chem. Soc. 1973, 95, 3310; Heathcock, JOC, 1980, 45, 1066; Mukaiyama JACS, 1974 96, 7503).

Note that the chemical shift of the CH(OH) proton is not in general a reliable indicator of stereochemistry, although for closely related series of compounds there might well be useful correlations.
This method will only work if the intramolecular hydrogen bonded conformations are the principal ones for both diastereomers. Thus, it sometimes fails in situations where the α and/or β-substituent is large, as in the α-t-Bu aldols below. Here gauche interactions destabilize the hydrogen bonded six-membered ring of the syn isomer, leading to a large coupling because of a high population of the non-hydrogen-bonded conformation with t-Bu and Ph anti periplanar in the syn isomer (Heng, Simpson, Smith J. Org. Chem. 1981, 46, 2932). Similarly, in more complicated systems additional conformational constraints can overwhelm the hydrogen bond effect. For example a 3-alkyl substituent in a cyclohexanone aldol has Jsyn > Janti (Kitamura, Nakano, Miki, Okada, Noyori J. Am. Chem. Soc. 2001, 123, 8939).

For use of 13C shifts to assign stereochemistry see: Heathcock, J. Org. Chem., 1979, 4294.
Felkin-anti-Felkin Stereochemistry of Aldol Adducts. A related analysis (Roush, et al. J. Org. Chem. 2002, 67, 4284-4289 DOI: 10.1021/jo0164148) led to a reliable method for assignment of stereochemistry to aldol products of methyl ketones such as the ones shown. In many cases, the downfield proton Ha for the Felkin diastereomer has larger Jax and a smaller Jbx, whereas for the anti-Felkin isomer the opposite pattern was observed - a smaller Jax and a larger Jbx. This trend is rationalized by the hydrogen-bonded conformations shown. The downfield shift of Ha in each isomer is presumably a consequence of the pseudo 1,3-diaxial interaction (green arrows) of this proton with the Me group in the expected conformations.

There are also some consistent empirical chemical shift effects for Ha and Hb.
Conformations of CH2 Chains. Adjacent CH2 groups in acyclic molecules (R1-CH2-CH2-R2) typically show apparent triplets, or higher multiplets if R1 and/or R2 contain vicinal protons coupled to the CH2 groups. These are actually AA'BB' or AA'XX' systems, and thus are inherently non-first order. It turns out that if R1 and R2 are sterically small, then the gauche conformation is sufficiently populated (anti/gauche ca 3:1) that nearly equal JAX and JAX' are seen, leading to the apparent triplets. If R1 and/or R2 is sterically large, then more complicated patterns are seen. See Sect. 5.15.
Exercise: Interpret the NMR spectrum below, assign all protons, and extract coupling constants. In particular, make sure you understand the multiplets at δ 3.2, 3.8 and 4.5. Explain the two doublets at δ 2.6 and 2.9. Consider possible conformations that might rationalize the couplings observed, as well as the chemical shift of the proton at δ 12.4.

5-HMR-5.7 Allylic 3J
Couplings of vinyl hydrogens to vicinal protons across single bonds (C=CH-CH) follow Karplus relationships similar to those of other vicinal couplings. The size of J is maximal at dihedral angles of 180° and 0°, and minimal when the C-H bonds are perpendicular (Θ = 90°), although the coupling does not go to 0.

In acyclic systems without strong conformational restrictions, rotational averaging produces couplings of 5-8 Hz, very similar to those observed in aliphatic chains. Examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17.
For cyclic olefins, the 3J coupling decreases as the ring size gets smaller. In cyclohexenes the couplings of an adjacent CH2 group to the vinyl hydrogens are typically 4-5 Hz for the equatorial H, and 1-3 Hz for the axial H, as shown in the figure above. In cyclohexene itself the average of these is observed. Examples: 1, 2, 3, 4, 5, 6.

Dienes: The central 3J coupling in acyclic dienes is typically 10 Hz, very similar to the 3Jcis across double bonds, provided that steric effects do not prevent the diene from achieving a near planar conformation. The coupling is reduced in cyclic dienes, both because the dihedral angle is now 0° instead of 180°, and because of inherent reduction in the coupling because of angle distortions. Examples: 1, 2, 3.

Aldehydes: In unconjugated aldehydes the 3J coupling is typically small (1-3 Hz). Examples: 1, 2, 3. The coupling becomes considerably larger in conjugated aldehydes like acrolein, where the dihedral angle will be either 0° or 180° to maximize overlap of the π systems. Examples: 1, 2, 3.

5-HMR-5.8 Olefinic Stereochemistry using 3JHH
The trans and cis couplings across a double bond are very reliable indicators of stereochemistry. With virtually no exceptions, 3Jtrans > 3Jcis, typical values are 17 and 10 Hz. However, the ranges do overlap for strongly electropositive substituents (J increases) and electronegative substituents (J decreases) on the double bond. Examples - E/Z isomers: 1, 2, 3, 4, 5, 6, 7, 8. Isolated Vinyl groups: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18; Allyl groups: 1, 2, 3, 4, 5, 6, 7. Miscellaneous: 1, 2, 3, 4, 5, 6.

The coupling varies with π bond order. Thus the cis coupling in benzene and other aromatic six and larger membered rings is typically below 10 Hz (one empirical equation is: 3J = 8.65·(π bond order) + 1.66):

The tropone shows larger bond-alternation effects than the aromatic tropylium ion or azulene.

Cycloalkenes smaller than cyclohexene show substantially reduced 3J values (Chem. Rev. 1977, 77, 599). Thus cyclopentenes can be easily distinguished from cyclohexenes and larger rings if this coupling can be identified. Examples - Cycloheptenes and larger rings: 1, 2, 3. Cyclohexenes: 1, 2, 3, 4, 5, 6, 7, 8. Cyclopentenes: 1, 2, 3, 4, 5, 6, 7.

Unsaturated O- and N-heterocycles also generally have smaller 3J values than hydrocarbon systems. Examples: 1, 2, 3, 4, 5, 6, 7.




Exercise: Analyze the proton NMR spectrum of an E / Z mixture of 1,3-dichloropropenes. Assign all peaks and determine J values. What is the isomer ratio? Click spectrum for answer.

Exercise: Assign all of the protons in the 1H NMR spectrum of methyl piperate. Click spectrum for answer.

Next Section: Long-range Coupling · Previous Section: Two-bond Coupling · Home