5-HMR-6 Long-Range (4J and higher) Proton-Proton Couplings
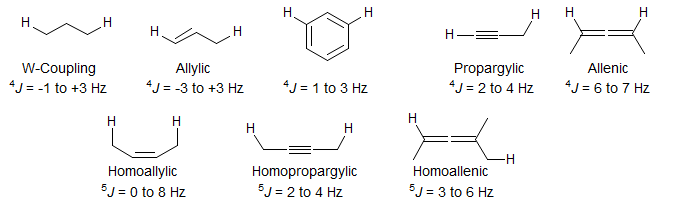
Proton-proton couplings over more than three bonds are usually too small to detect easily (< 1 Hz). However, there are a number of important environments where such couplings are present, and can provide useful structural information. Coupling across π-systems are the most frequently encountered 4J couplings: the meta-coupling in aromatic compounds, and the 4-bond allylic, propargylic and allenic couplings. 4-Bond couplings across saturated carbons (sp3) or heteroatoms are rarer, and are usually seen only in cyclic compounds when there is a favorable geometric alignment along the H-C-C-C-H chain ("W-Coupling"). Longer range couplings (5J and higher) are also observed, particularly in acetylenes and allenes (Chem. Rev. 1977, 77, 599).
5-HMR-6.1 W-Coupling in Saturated Systems 4J
Normally long-range couplings across saturated carbons (or O and N) are too small to detect easily (<1 Hz). However, if there is proper orbital alignment between C-H bonds and the intervening C-C bonds then 4-bond and higher couplings can be observed. The most favorable alignment is the W arrangement of the connecting bonds ("W-coupling"), in which the H-C-C and C-C-H fragments are close to coplanar in an anti-arrangement. However, couplings across U-shaped and sickle-shaped coplanar HCCCH fragments can also sometimes be detected. Long-range couplings can become quite large in rigid strained bicyclic ring systems and/or when there are multiple coupling pathways available.
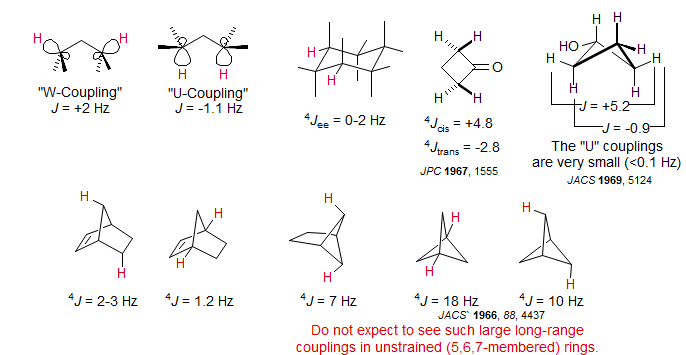
Cyclohexanes. The 1,3-equatorial protons in cyclohexanes have an ideal W-arrangement, and such coupling is frequently seen. Examples: 1, 2, 3, 4.
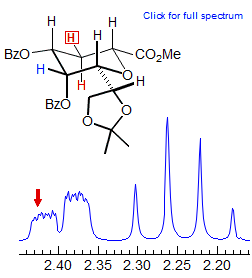
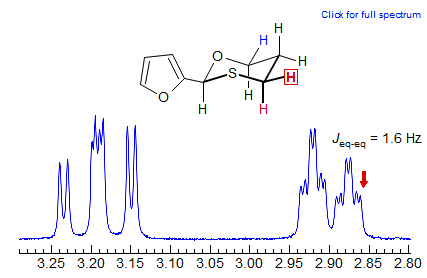
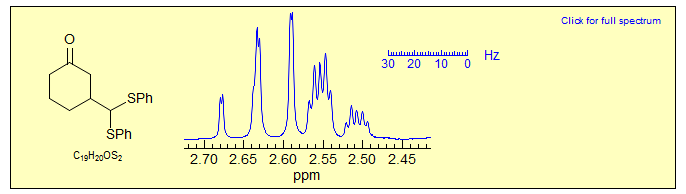
Exercise: Examine the 300 MHz spectrum of two of the protons of 3-bis(phenylthio)methylcyclohexanone reproduced below. Assign the protons and analyze the couplings (Sikorski/Reich).
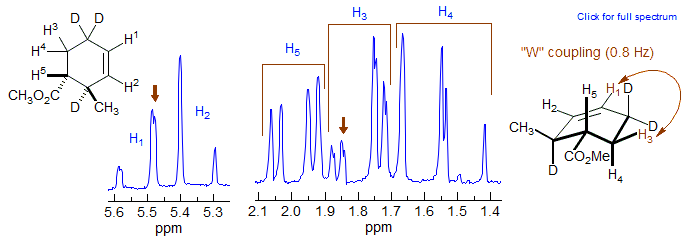
Vinyl protons in cyclohexenes also have a W-relationship with equatorial protons 2 carbons removed, and usually show significant coupling (0.8 Hz in the example below). For other examples see: 1, 2.
W-Coupling is also seen in cyclopentanes. Example: 1.
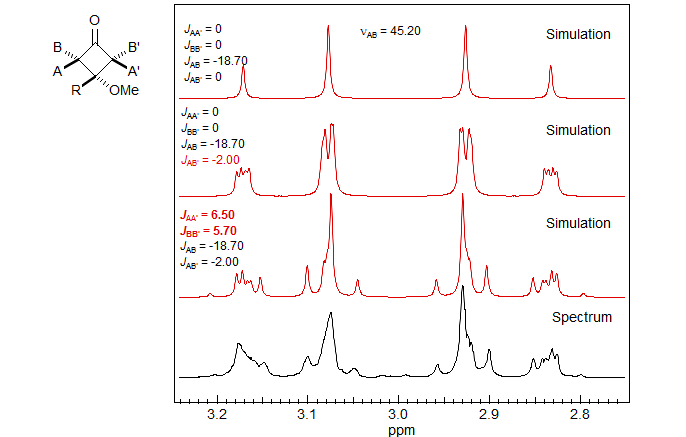
Cyclobutanes generally show substantial cross-ring 4J couplings, with 4Jcis, which has the proper orientation for a W-coupling, greater than 4Jtrans. In fact, Jcis > 0 and Jtrans < 0 in almost all cases (A. Gamba, R. Mondelli Tetrahedron Lett. 1971, 2133), so this coupling can be used to assign stereochemistry in cyclobutanes. The figure below illustrates the effect of the long range couplings in a cyclobutanone.
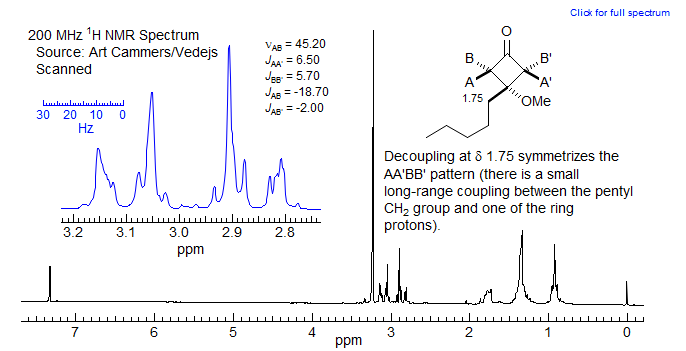
A simple AB quartet would be expected if there were no long-range couplings (top simulation). Clearly, that isnot what was observed. An AA'BB' simulation gives the parameters shown on the figure. The pattern is not completely centrosymmetric because there is a small long-range coupling from the side-chain CH2 (δ 1.75) to one of the cyclobutane protons.
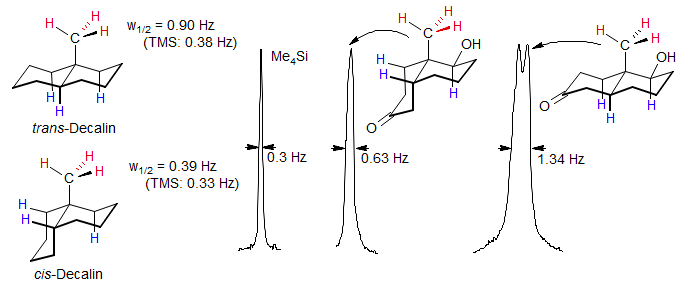
Stereochemistry of cis/trans Decalins. A useful application of long range couplings for the assignment of ring-fusion stereochemistry in decalin ring system bearing an angular methyl group has been developed (Williamson, K. L.; Howell, T.; Spencer, T. A. J. Am. Chem. Soc. 1966, 88, 325). In the trans-decalins, there are usually several ideal W-pathways for long range coupling between the methyl group and axial protons. In cis-decalins, there are fewer or no such pathways. Thus in a pair of cis/trans isomers, the methyl group in the trans isomer will usually be broader (or will actually show splitting), whereas the cis isomer will have a sharper (unsplit) methyl group.
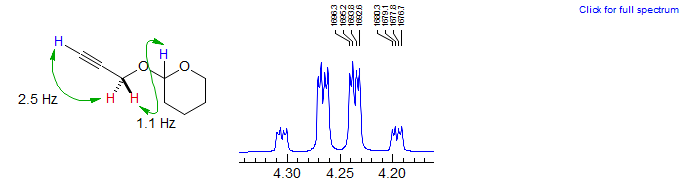
W-Coupling Across Heteroatoms. A small 4J across oxygen was observed in the THP-protected propargyl alcohol shown below. The CH2-O protons indicated are coupled both to the alkynyl proton (4J = 2.5 Hz) and the methyne of the acetal (4J = 1.1 Hz)
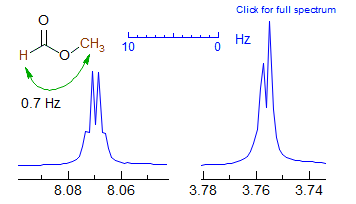
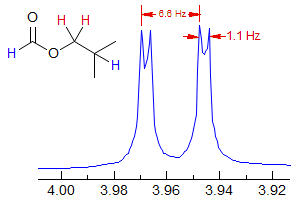
Formate esters usually show a 4J coupling from the formyl to the OCHn proton, as seen in the spectra of methyl and isobutyl formate below.
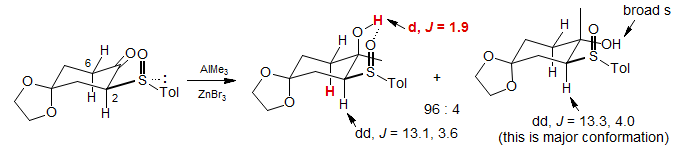
In conformationally well-defined systems significant 4J couplings can be seen to OH and other XH protons. In the example below, the long-range W-coupling between the OH proton and the axial proton at C6 was used to assign configuration to the major isomer formed in the reaction. In the minor isomer the OH proton was not detectably coupled. The well-defined cyclohexane 3Jax-ax and 3Jax-eq at C2 in both isomers shows that the ring-flip isomer shown predominates (Bueno, A. B.; Carreno, M. C.; Ruano, J. L. G Tetrahedron Lett. 1995, 36, 3737).
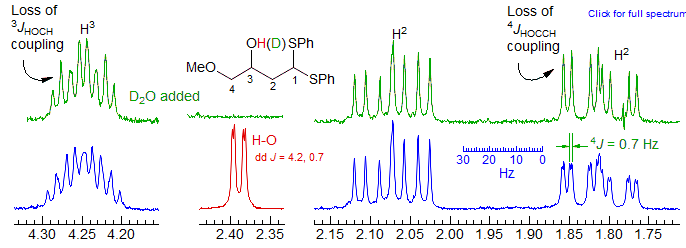
Both 3JHOCC (to the H3 proton) and 4JHOCCC (to one of the diastereotopic H2 protons) was detected in the alcohol shown below. Both couplings essentially disappear when the sample was shaken with D2O.
In a related system, the observation of an unusually large 4J across the sulfone sulfur was interpreted in terms of the conformation shown, in which the methyl group is over the ring, rather than alternative conformations in which the sulfone oxygen is over the ring (Kaloustian, M. K.; Dennis, N.; Mager, S.; Evans, S. A.; Alcudia, F. Eliel, E. L. J. Am. Chem. Soc. 1976, 98, 956-965).
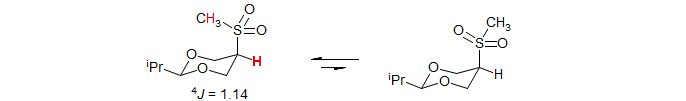
5-HMR-6.2 Allylic Coupling 4J
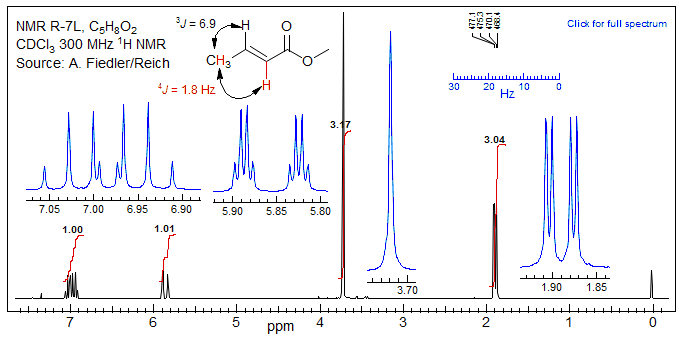
4-Bond coupling of vinyl to allylic hydrogens is usually easily observable. A typical example is methyl crotonate, where the 4J between the CH3 and the α-CH is 1.8 Hz.
We can think of the allylic 4J coupling as having two components, the usual W-coupling transmitted through the σ-system, which is positive and is maximized for the trans proton when the allylic C-H bond is in the plane of the vinyl C-H group (Θ = 0 °, J > 0), and a π-component, which is negative, and whose magnitude is maximized when the allylic C-H bond is perpendicular to the double bond (Θ = 90 °, J < 0) (Garbisch, J. Am. Chem. Soc. 1964, 86, 5561). The positive σ-contribution added to the larger negative π-contribution normally results in a numerically slightly smaller (negative) coupling to the trans vinyl proton, but the effect is small, and not reliable enough for the unambiguous determination of double bond stereochemistry (note the marked entry below in which the magnitude of Jtrans > Jcis) (Barfield, M.; Chakrabarti, B. Chem. Rev. 1969, 69, 757). Examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34.
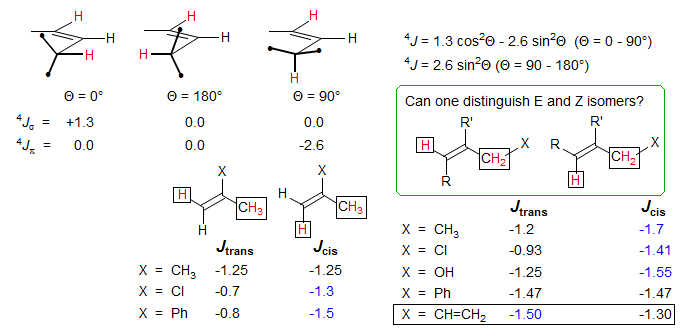
In special cases, the 4J allylic coupling can be quite different for cis and trans protons, for example in the exo-methylene cycloheptanone below the 4Jtrans is not detectable (probably less than 0.3 Hz, assigned on the basis of the downfield chemical shift) whereas the 4Jcis is 1.1 Hz
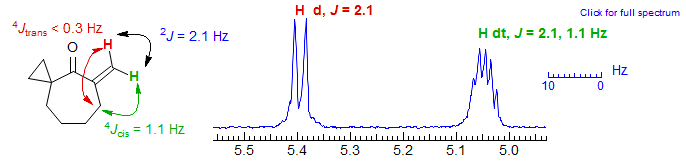
It is not uncommon in cyclic alkenes for the magnitude of 4J and 3J allylic coupling to be nearly the same, since the alignment for 4J is often optimal (C-H perpendicular to π system for pseudoaxial protons), whereas the alignment for 3J is poor. An example is the cyclohexadiene below, where 4J is 2.6 Hz and 3J is 3.3 Hz. Other Examples: 1, 2.
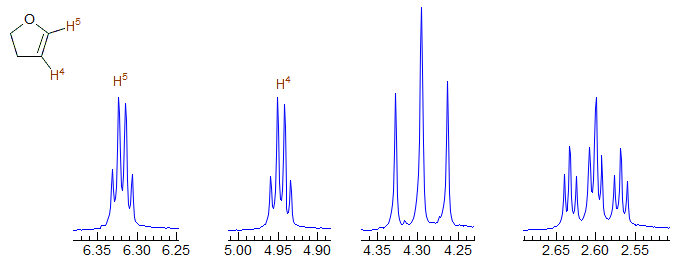
In fact, in 2,3-dihydrofuran, the two couplings are identical within experimental error - both vinyl protons are apparent quartets.
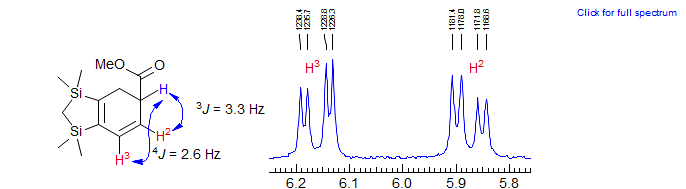
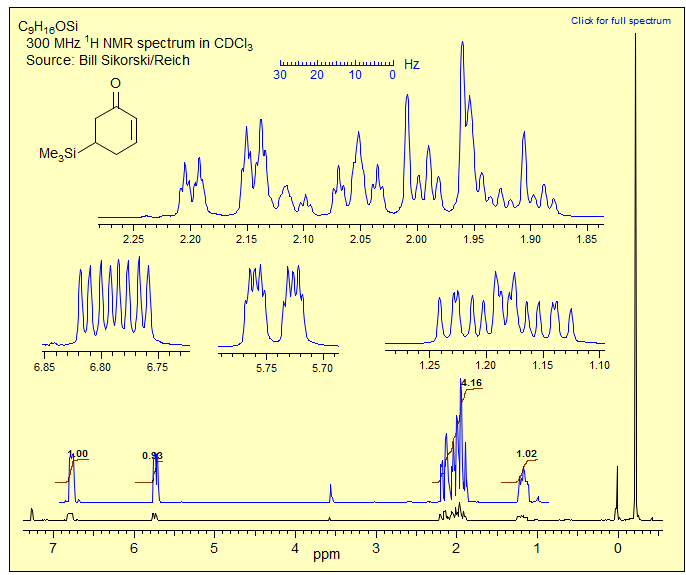
Exercise: If you have learned the skills of handling first order multiplets and know enough about the size of 2J, 3J and 4J H-H coupling constants, you should be able to analyze the multiplets in the NMR spectrum of 5-trimethylsilyl-2-cyclohexenone. Apart from the usual leaning, all of the multiplets are basically first order, and only a few of the peaks are badly overlapped. Identify all couplings and assign the protons. Begin by drawing a proper conformation of the molecule, and assigning and analyzing the isolated multiplets. Then tackle the overlapped region δ 1.8-2.2.Click the spectrum for answer
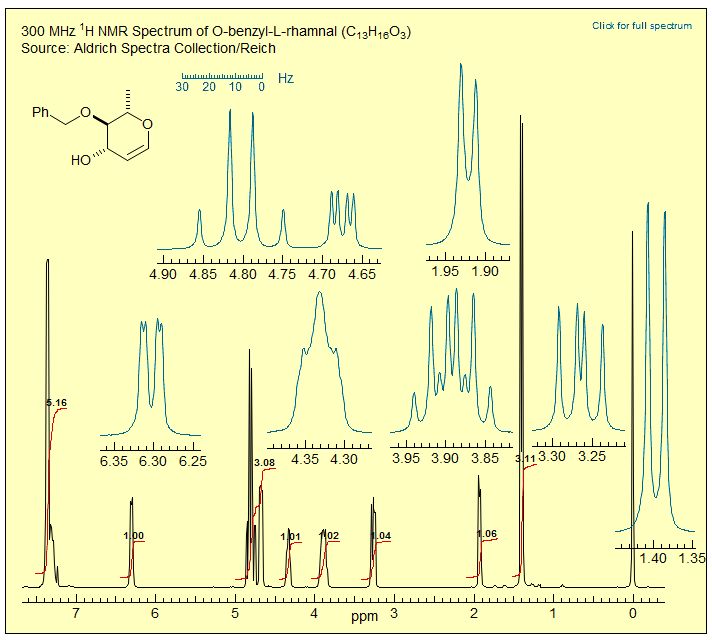
Exercise: Examine the 300 MHz 1H NMR spectrum of O-benzyl rhamnal below. Assign all protons and extract couplings.
5-HMR-6.3 Meta and Para Coupling in Aromatic Rings 4J, 5J
4-Bond coupling between 1,3-related protons in aromatic compounds (typically 1.5-3 Hz) is almost universally seen and, together with the larger (6-8 Hz) ortho-coupling, often provides a method of securely identifying the positions of substituents. For examples see NMR Spectra Gallery - Aromatics.
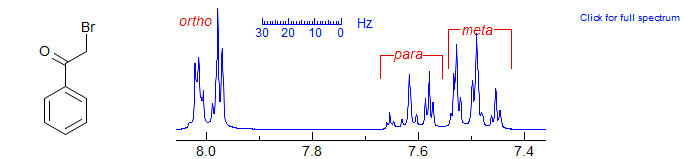
Monosubstituted benzenes form an AA'BB'C pattern, which is never first order, although when the shifts are well separated the pattern has the rough appearance of a d of multiplets (ortho), tm (meta) and tm (para). See here for an analysis. Examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14.
Disubstituted benzenes or monosubstituted pyridines: the three isomers are very distinctive, as can be seen below. 1,2-Disubstituted benzenes have the 3 and 6 protons coupled ortho and meta (dd) and the 4,5 protons as triplets of doublets (as in the example below), or ddd if the two ortho couplings are different. Examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14.
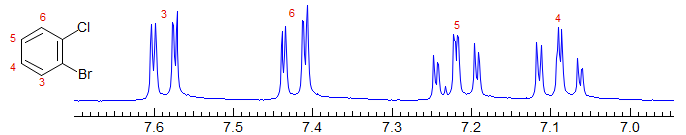
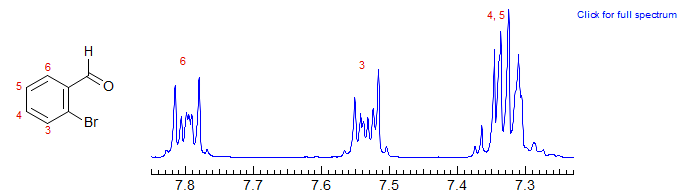
Of course, the pattern above is all for well-separated chemical shifts for non-equivalent protons. If any of the coupled protons becomes superimposed, particularly protons ortho to each other (larger coupling) the spectra quickly become very complicated. Compare 2-bromobenzaldehyde below with 2-bromochlorobenzene above:
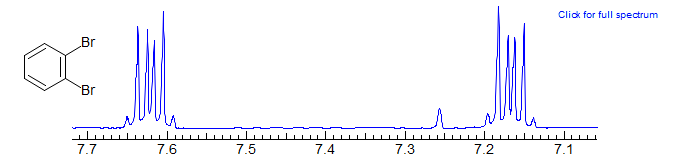
If the two substituents are identical, then the spectrum is alway complex, an AA'BB' pattern Examples: 1, 2, 3.
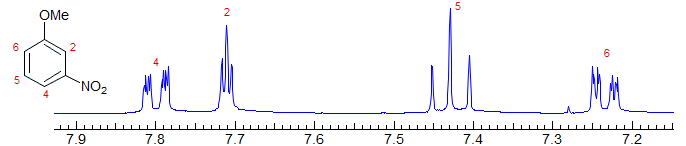
1,3-Disubstituted benzenes have the 2-proton as a narrow triplet or dd with nearly equal J values (2 meta couplings), the 5-proton as a wide triplet (or dd) from two ortho couplings, and the 5,6-protons as dt or ddd from one ortho and two meta couplings (which are different in the example below because of the large electronic difference between the substituents). Examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.
When the substituents are identical, the pattern is simpler (A2MX). Examples: 1, 2, 3, 4.
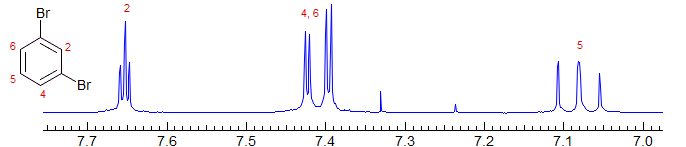
Superposition of coupled protons leads to interpretation problems. Compare 3-bromoanisole below with 3-nitroanisole above:
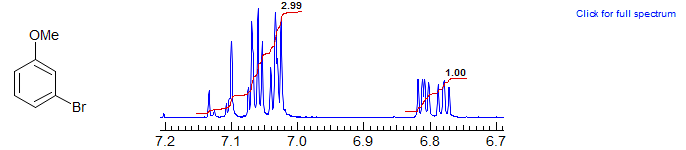
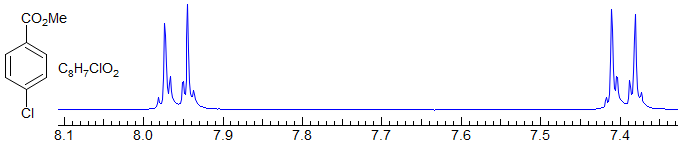
1,4-Disubstituted benzenes are never first order (they are an AA'BB' system), but they do have a strong doublet as a dominant feature resulting from the main coupling present, that between the two ortho protons. The line separation is close in magnitude to the ortho coupling, but not identical to it (the line separation is Jortho + Jpara). Examples: 1, 2, 3, 4, 5, 6, 7, 8, 9.
There are three position isomers of trisubstituted benzenes - 1,2,3- 1,2,4- and 1,3,5-. In each case the NMR spectrum (if sufficiently first order) fully identifies the substitution pattern. Distinguishing various ways of placing substituents at these positions may not be as easy, since this requires careful analysis of chemical shifts. Examples - 3 isomers: 1, 2, 3; 1,2,3-substitution: 1, 2, 3, 4 1,2,4-substitution: 1, 2, 3, 4, 5, 6, 7, 8. Simpler AX2 or AB2 spectra are seen for symmetric 1,3,5- (t and d with J ≈ 2 Hz) and 1,2,3-isomers (t and d with J ≈ 8 Hz). Examples: 1, 2, 3. For quinone examples see: 1, 2.
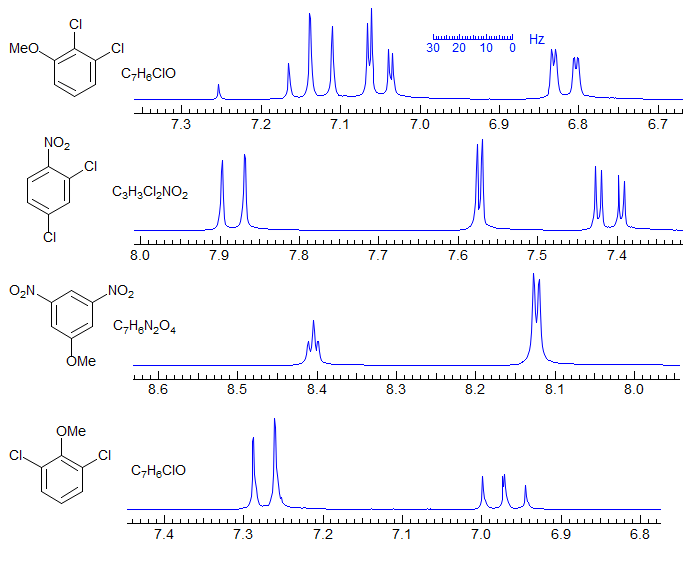
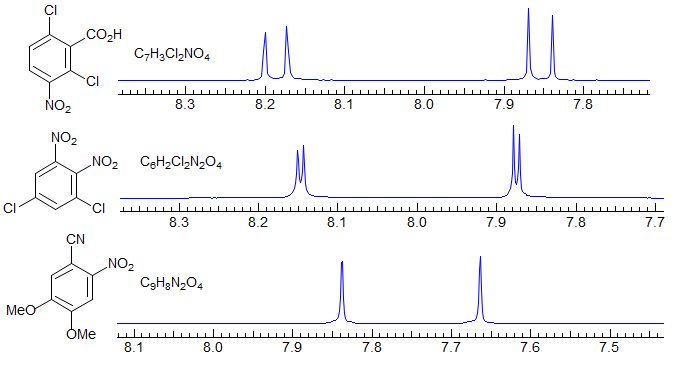
Simplest are tetra-substituted benzenes (or trisubstituted pyridines) where the two remaining protons are either an AB quartet with J around 7 Hz (for ortho), 2 Hz (for meta) or <1 Hz (for para), essentially two singlets. Examples: 1.
Naphthalenes: The considerations above can be used in identifying substitution patterns for a host of other aromatic systems. Consider the disubstituted (I, OMe) naphthalene below:
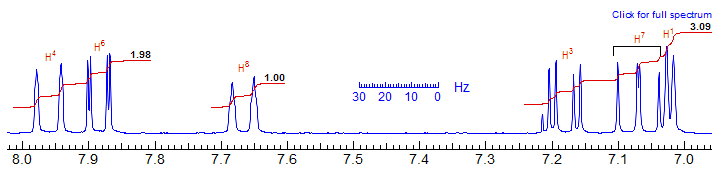
The dd at δ 7.07, J = 8, 7.5 Hz means that one ring has three adjacent protons (6,7,8 protons), so one group must be at the α-position. The d (J = 2.5) at 7.03 matches up with the dd (J = 9, 2.5) at 7.19. This requires that the other ring has 1,3,4-proton substitution, so the second substituent must be at the β position. This should be the OMe group, because of the upfield shift (δ 7.03, 7.17) of the two protons ortho to it. This leads to a preference for 1 and 2 of the four possible structures shown. Deciding among 1 and 2 is more difficult and requires a more careful analysis of chemical shifts - the actual structure is 1. Additional examples: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16.
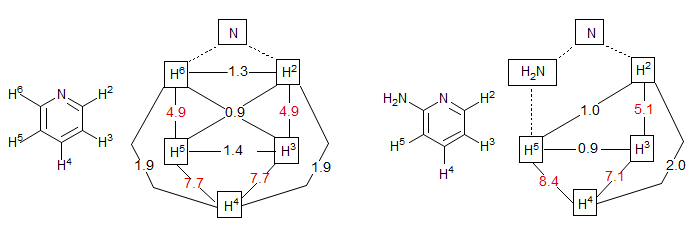
Pyridines: The couplings in pyridines are more or less like those in benzenes, but with 3J2-3 generally a little smaller (5-6 Hz) than those in benzenes, and 3J3-4 larger (7-9 Hz). The numbers for pyridine itself (Castellano, Sun, Kostelnik J. Chem. Phys. 1967, 46, 327) and for 2-aminopyridine are shown below. Examples: 1, 2, 3, 4.
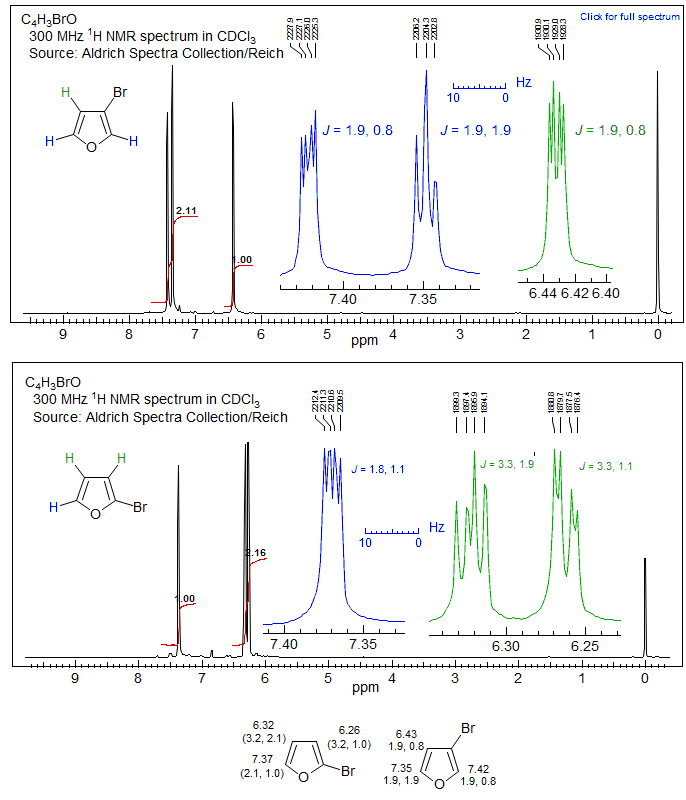
Furans: In five-membered aromatic heterocycles (furan, pyrrhole, thiophene, etc) the coupling constants are less distinct than in the 6-ring aromatics, with 3J much smaller, usually < 5 Hz, and the 4J couplings not much smaller than the 3J ones (1-3 Hz) . This is illustrated in the two furans below, where the very distinct chemical shifts of the 2 and 3 protons is a simpler diagnostic for the position of the substituents than is provide by the coupling. Other examples: 1, 2, 3, 4, 5.
5-HMR-6.4 Benzylic Coupling 4J
Coupling between benzyl protons and ortho (and even meta and para) hydrogens is generally detectable. The coupling is related to π-bond order, so it is usually smaller than allylic coupling, typically <1 Hz. It is thus often not resolved, but almost always causes significant broadening of both the aromatic and benzyl protons. This can be clearly seen in the spectrum of p-methylacetophenone below, where the Ar-CH3 is wider and thus noticeably shorter than the C(=O)-CH3, and the protons ortho to the CH3 are broader than those ortho to the acetyl group. For other examples of this effect see: 1, 2.
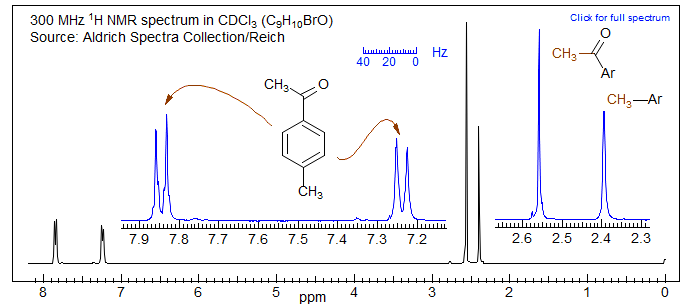
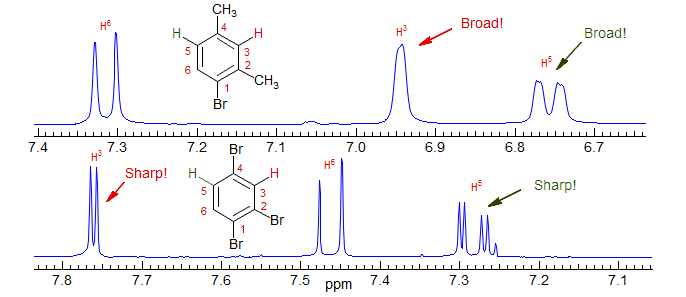
Note the much broader aromatic peaks (almost no meta-coupling detectable) in the bromoxylene than in the tribromide due to coupling of the aromatic C-H to the CH3 protons.
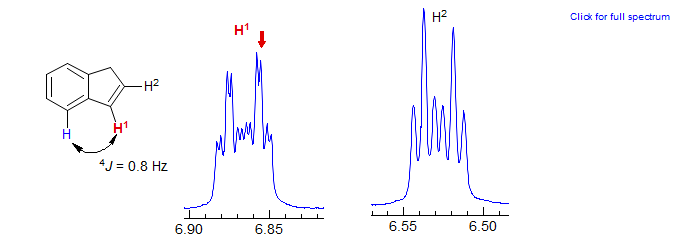
In favorable cases, such as indene below, the 4J benzylic coupling can be easily observed: Other Examples: 1 (thiophene).
5-HMR-6.5 Homoallylic Coupling 5J
Couplings across 5 bonds are unusual, but can be seen under favorable circumstances. Coupling is optimal when both C-H bonds are aligned with the π-orbital of an intervening double bond (perpendicular to the plane of the double bond). Especially large long-range couplings are seen for 1,4-cyclohexadienes and related structures where there are two paths for the coupling.
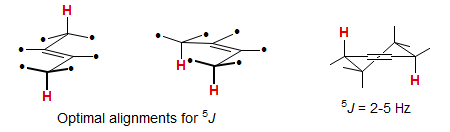
Some extreme examples of large homoallylic coupling:
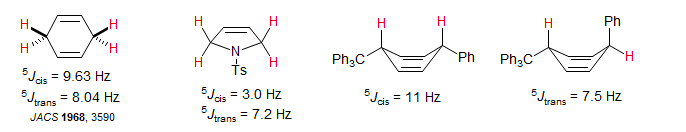
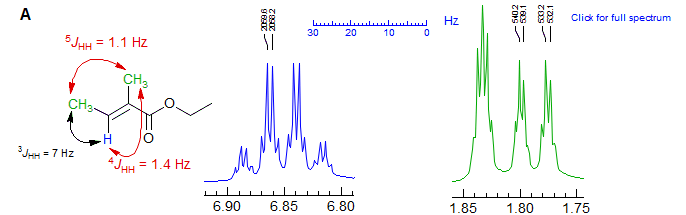
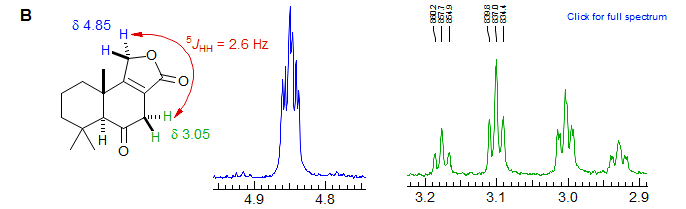
Typical examples of 5J homoallylic coupling are shown below. Spectrum A also shows 4J allylic coupling. Other Examples: 1
5-HMR-6.6 Long range Couplings in Acetylenes and Allenes
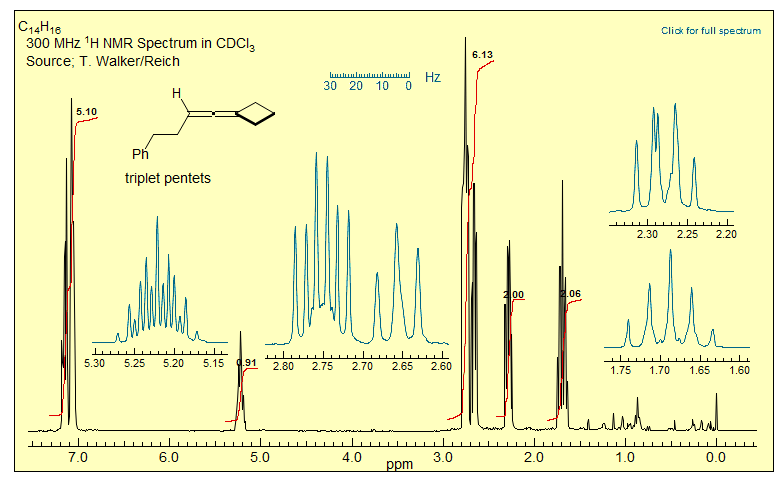
No special structural features are required to observe 4- and 5-bond couplings across acetylenes and allenes - such couplings are always present. Even couplings across 6 and more bonds are detected across polyacetylene or cumulene chains. Examples alkynes: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18. Examples allenes: 1, 2.
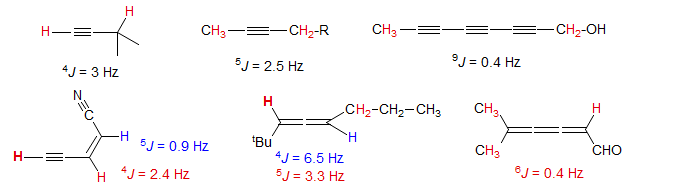
Long range couplings like this are also observed across nitrogen as in the nitrilium ion below:

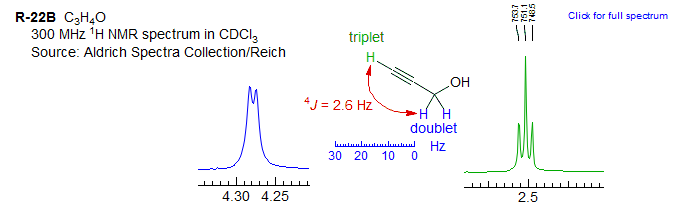
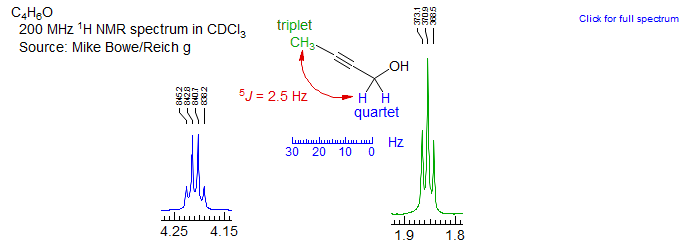
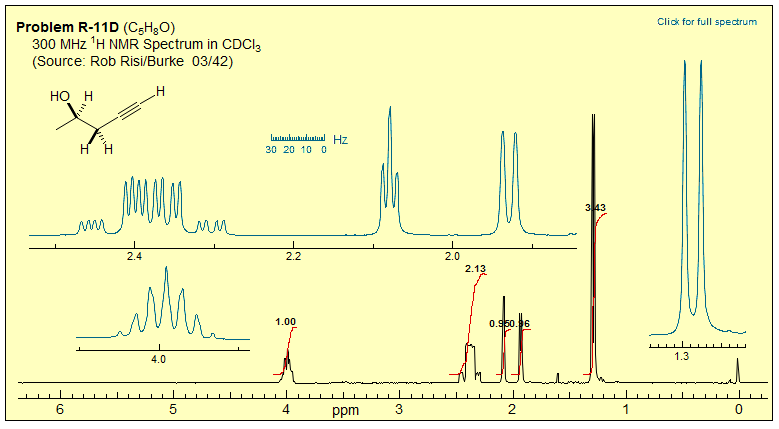
Exercise: Analyze the NMR spectra below.
Next Section: Pople Spin Nomenclature · Previous Section: Three-bond Coupling · Home