6-CMR-1 Measuring 13C NMR Spectra
The low natural abundance of the NMR active isotope of carbon, 13C, makes the measurement and interpretation of carbon NMR spectra a considerably different process than measurement and analysis of proton NMR spectra.
13C6 I = ½ Natural abundance: 1.1%
12C6 I = 0 Natural abundance: 98.9%
Spectrometer Frequency:
6-CMR-1.1 Sensitivity
The low natural abundance of 13C has three principal consequences:
1. It can be much more time-consuming to obtain 13C than 1H NMR spectra, especially if quantities or solubility is limited. Whereas an 1H spectrum on 1 mg of a typical organic compound can usually be obtained in 15-30 minutes of spectrometer time, it might take several hours to obtain a much lower quality 13C spectrum on the same sample. This is because, in addition to the 1% natural abundance of 13C (vs >99% for 1H), the Boltzman population difference between the α and β states is smaller (the energy difference is only 1/4 as large), and the weaker magnetic dipole makes signals inherently harder to detect. Thus, unless your compound does not have useful 1H signals, you will usually measure many more 1H than 13C NMR spectra.
Inherent sensitivity: 1.59% (1H = 100). Sens. = (γC/γH)3
Actual sensitivity: 0.017% (1/5700)
2. The effects of 13C nuclei on spectra of other nuclei (e.g., 1H, 19F, 31P) are very minor. Each proton (or F, P) signal is surrounded by 13C satellites separated by 1JC-H (typically 120-150 Hz), each with an intensity of 0.5% of the central peak. There are also close in satellites from longer range couplings to carbon - 2JC-H and 3JC-H, typically 2-15 Hz. The central peak arises from the 98.9% of 12C which is NMR transparent. The inner 13C satellites are often hard to resolve from the central peak(s), but the outer ones can be readily detected for sharp peaks. In addition to being useful for measuring proton-carbon couplings, these satellites can sometimes be used for measurement of JHH between equivalent protons. They are also useful as integration standards for quantifying the amount of minor isomers or other impurities.
3. Coupling between carbons (JCC) is not usually observed, because two adjacent 13C nuclei occur in only 1.1% of the carbons. There are thus 13C satellites on the carbon peaks (each about 0.5% of the intensity of the main peak), in the same way that there are 13C satellites on proton spectra. The couplings can be measured directly with some difficulty by accumulating many scans on a very concentrated sample, but a better way is to use one of the multi-pulse 2D experiments (e.g., INADEQUATE) which nulls the central peaks due to adjacent 12C atoms. Even so, large samples and long acquisition times are required.
6-CMR-1.2 Decoupling
Most 13C NMR spectra are very complex. The methyl carbon of an ethoxy group will appear as a large quartet, with each line further split into triplets. Even in fairly simple molecules each carbon may be coupled to a number of different protons. In complicated molecules, these multiplets overlap badly, and may be impossible to analyze.
1JCH = 100-250 Hz
2,3JCH = 2-10 Hz
To simplify 13C spectra, we usually use some form of broadband decoupling (noise decoupling) to remove the effect of proton couplings. This also dramatically increases signal intensity, since now all carbons appear as singlets (assuming absence of other spin 1/2 nuclei like 31P or 19F). The increase is actually greater by a factor of 2-3 than would be predicted on the basis of simply combining the 13C multiplet intensities because the Nuclear Overhauser Effect causes additional increases in signal intensity. More about this in Section 8.2.
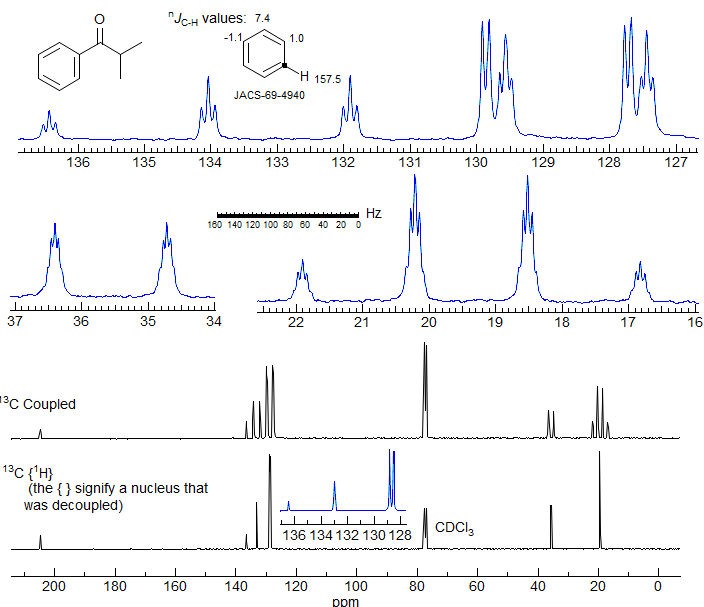
Figure 6-1.1. 75.4 MHz 13C NMR spectrum of isobutyrophenone in CDCl3. Identify all of the C-H couplings in the expansions of the coupled spectrum. Assume the coupling within the aromatic ring follow those given for benzene itself. Note that the very closely spaced ortho and meta carbons can be assigned based on the 3JCH couplings observed -- the dd corresponds to the meta carbon (it has only one meta proton), the dt to the ortho carbon.
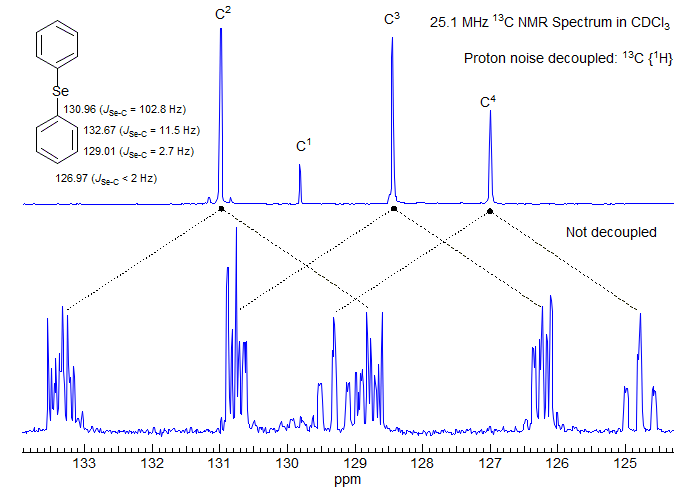
Figure 6-1.2. 25 MHz 13C NMR spectra of diphenyl selenide in CDCl3.
Figure 6-1.2 shows the fully coupled and decoupled 13C NMR spectra of diphenyl selenide. Although the large 1JC-H splittings are easy to identify, the fine structure of the individual multiplets is not first order (e.g., only the para carbon has an approximately centrosymmetric pattern, the others do not). This is because we are looking at the X part of an AA'BB'CX pattern (ABC are protons, X is carbon). Since the AA' and BB' parts are strongly coupled, we see the usual complex effects of "virtual coupling" on the X resonance (see Section 5-15, Section 5-16). When noise 1H decoupling is applied, the spectrum becomes much more intense, and only 4 lines remain, one for each carbon.
In this compound we have a second magnetically active nucleus (77Se, natural abundance 7.5%, I = 1/2), so each of the 13C peaks has 77Se satellites, although coupling between C-4 and the selenium is too small to detect (the satellites are under the main peak).
6-CMR-1.3 Attached Proton Tests
The disadvantage of obtaining fully decoupled spectra is that all information about how many protons are coupled to each carbon is lost. Since information about the number and chemical shifts of C, CH, CH2 and CH3 carbons is valuable in making structure and spectral assignments, several types of experiments to determine 13C multiplicities have been developed (fully coupled spectra are not usually used because of extensive signal overlap and poor signal-to-noise).
Single Frequency Off-Resonance Decoupling (SFORD).
The oldest method for determining attached protons is the SFORD technique, now replaced by more efficient methods. It involves application of a powerful single frequency proton decoupling 500-2000 Hz upfield or downfield of the proton chemical shifts in the sample. All C-H couplings are reduced in proportion to:
1. The distance (Δδ) from the decoupler frequency to the proton position,
2. The power of the decoupler.
Typical experiments would reduce the normal one-bond couplings of 125-250 Hz to perhaps 12-20 Hz, and the longer range couplings to <1 Hz. The spectrum now appears as a series of singlets, doublets, triplets and quartets depending on whether the carbon is quaternary, or has one, two or three protons attached. Fig. 6-1.3 illustrates the effect of decoupler offset on the 13C NMR spectrum of methanol.
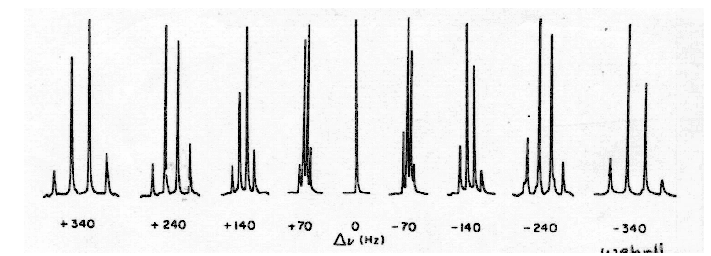
Figure 6-1.3. Methyl carbon quartet of methanol obtained for various offsets of the decoupler frequency from the 1H chemical shift of methanol (from Wehrli and Wirthlin).
Some difficulties with this technique are:
1. Sensitivity is not very good.
2. Multiplets are not always clean.
3. For complex molecules, some regions of the spectrum can become hopelessly crowded.
6-CMR-1.4 Spectral Editing
There are a number of multi-pulse experiments which render the signals in a 13C NMR spectrum with positive and negative intensities (sometimes zero) according to the number of attached protons.
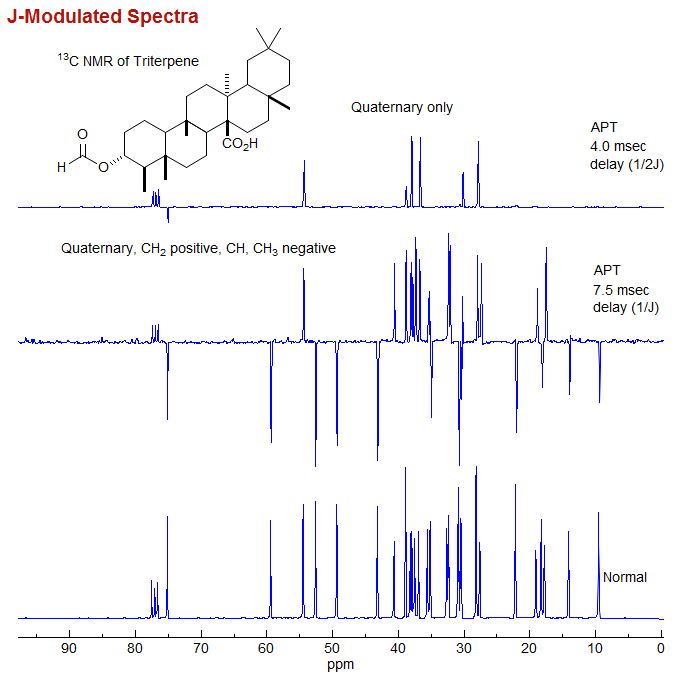
J-Modulated Spectra. This is the most primitive form of spectral editing. By placing a suitable delay time between the pulse and the beginning of the acquisition, spectra are obtained in which C and CH2 groups are positive, and CH and CH3 are negative.
In this experiment, after the pulse there is a short delay, during which the decoupler is turned off, and the 13C NMR spectrum becomes modulated by the CH coupling frequency. After the delay the decoupler is turned on, and the FID is recorded. If the delay is 1/J then the quaternary and CH2 carbons are positive, and the CH and CH3 signals are negative. If the delay is 1/2J all peaks except quaternary are nulled. Of course, experiments of this type will only work properly if the JC-H are the same for all carbons, which is not always the case, leading to some artifacts when the coupling is unusually large, as for C≡C-H. More on this experiment in Sect 8-Tech-9.1.
DEPT (Distortionless Enhancement of Polarization Transfer): The DEPT technique has proven superior to others in providing information on attached protons reliably, efficiently and with high selectivity. It is a proton-carbon polarization transfer method, so DEPT spectra are actually more sensitive than normal acquisitions. A set of spectra with pulse delays adjusted for π/2 (DEPT-90) and 3π/4 (DEPT-135) are taken. The DEPT-90 spectrum shows only CH carbons, the DEPT-135 shows positive CH3 and CH, and negative CH2 signals. It is important to understand that the appearance of positive and negative signals can be reversed by phasing, so it is necessary to have some way of determining whether the spectrum has been phased for CH2 positive or negative. Quaternary carbons are invisible (Fig. 6-1.4).
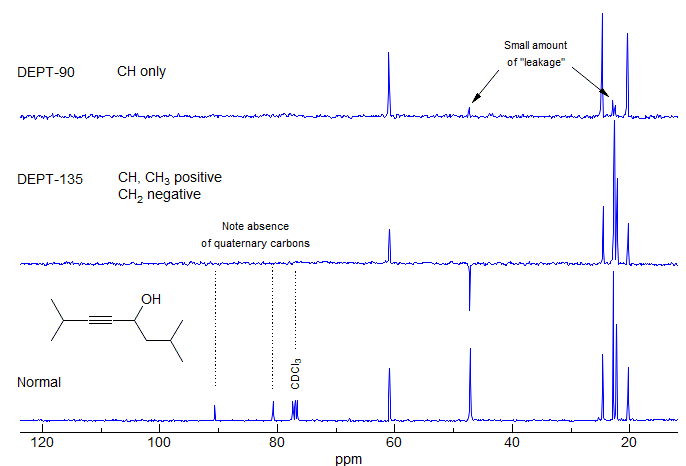
Figure 6-1.4. The normal 13C NMR spectrum and a typical set of DEPT spectra of an alkyne. Note the absence of the quaternary alkyne carbons in the DEPT spectra, and the presence of small peaks for the CH2 and CH3 signals in the DEPT-90 spectrum, which, in principle, should have only CH signals.
"Leakage" can occur in DEPT-90 spectra because 1JC-H varies as a function of environment, and the technique assumes that all 1JC-H are identical. This can result in small peaks for CH2 and CH3 signals, which should have zero intensity. For similar reasons the C-H of terminal acetylenes (C≡C-H) will show anomalous intensities in DEPT spectra (either nulled or very small in DEPT-90, or present in DEPT-135) because the C-H coupling is much larger (around 250 Hz) than the normal value of 125 Hz for which the DEPT experiment is usually parameterized. Of course, leakage can also result from an incorrectly calibrated pulse width for the spectrometer.
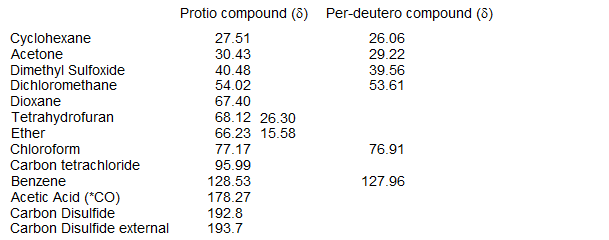
6-CMR-1.5 Referencing C-13 NMR Spectra
Tetramethylsilane (TMS) is the primary reference for C-13 spectra. The relatively low sensitivity of C-13 NMR requires the addition of substantial amounts of TMS, so it is common to use solvent peaks as a secondary reference. Below are listed chemical shifts of several common solvents used in NMR spectroscopy. Note that isotope shifts are quite large in C-13 NMR, so separate values are reported for the deuterated and protonated solvents (from Levy, J. Magn. Res. 1972, 6, 143; Reich unpublished work).
Compound (solvent)
Next Section: Origin of Shifts · Home