6-CMR-2 Origin of Chemical Shifts
For a tabulation of thousands of 13C chemical shifts see C-13 Shift data.
There are three principal effects which control NMR chemical shifts: diamagnetic, paramagnetic and neighboring group anisotropy:
Diamagnetic term - σd: electron circulation within an s orbital causes shifts to low frequency (upfield, shielding) of the local nucleus. This term dominates proton chemical shifts, it changes in a more or less predictable way with electron density and hybridization so that chemists feel comfortable in rationalizing many of the effects observed.
Neighboring group anisotropy - σn: diamagnetic circulation causes local magnetic fields, which will have an effect on neighboring nuclei. These effects are on the order of at most a few ppm, and are constant (same size in ppm) independent of the nucleus which is being affected. Thus, anisotropy shifts are important in proton NMR, where they are significant when compared with other effects, but are usually insignificant for nuclei (such as 13C) that have shift ranges of hundreds of ppm.
Paramagnetic term - σp: circulation of electrons between ground and excited states of p orbitals induced by the external magnetic field causes large high-frequency (downfield, deshielding) shifts of nearby nuclei. Although the forces exerted on electrons by the magnetic field are small compared to the energy differences between ground and exited states, the shifts are very large, and even minuscule amount of paramagnetic circulation cause very large chemical shifts. All nuclei heavier than 1H (with the possible exception of 6Li and 7Li) have σp as the principal chemical shift effect.
It is fortunate that σp often responds in the same way to electron density as does σd, since this results in parallel 1H and 13C chemical shift features. However, a qualitative description of the relationship between δ and molecular features must consider two factors which affect σp and σd quite differently and may cause very counterintuitive behavior: the 1/ΔE dependence, and orbital symmetry effects.
(1) Since we are dealing with promotion of electrons from ground to excited states, the energy separation between filled and empty orbitals (HOMO-LUMO separation) has an important effect, i.e. low-lying unoccupied molecular orbitals of the correct symmetry (see (2) below) result in large paramagnetic (high-frequency) shifts. The very unusual 1H shifts of anti-aromatic compounds results from the very small HOMO-LUMO energy gap in 4n π systems.
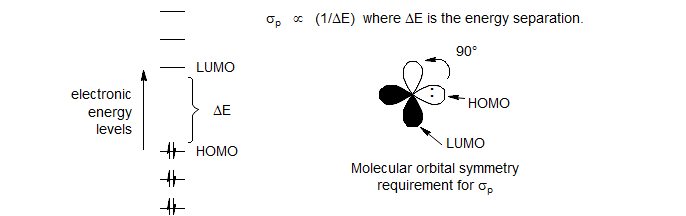
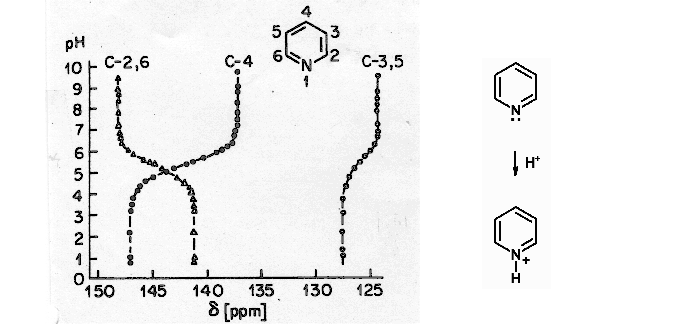
(2) The orbital symmetry relationship between the orbitals determines whether circulation between any pair of filled and empty orbitals can occur. Since the closest pair is the HOMO and LUMO, we restrict our consideration to these. In order for paramagnetic electron circulation (mixing between ground and excited states) to occur, the HOMO and LUMO orbitals must have the same symmetry after a 90° rotation. Thus if the HOMO and LUMO are p-orbitals which are perpendicular to each other, σp will be large. An especially striking example is provided by the N-protonation of pyridine (Breitmeier, Spohn Tetrahedron 1973, 29, 1145).The C-3,5 and C-4 shifts move to high frequency on protonation, as expected from the increase in positive charge at nitrogen. On the other hand, the C-2,6 shifts move to low frequency. The latter are close to the N-lone pair (the HOMO in the Figure above) which has a proper symmetry relationship with the π* orbitals of the aromatic ring, and are thus most affected by the reduction of paramagnetic electron circulation on protonation at nitrogen. The 15N chemical shift also moves to lower frequency on protonation (from -68 ppm for pyridine to -166.6 for pyridinium JACS 1978, 100, 4969).
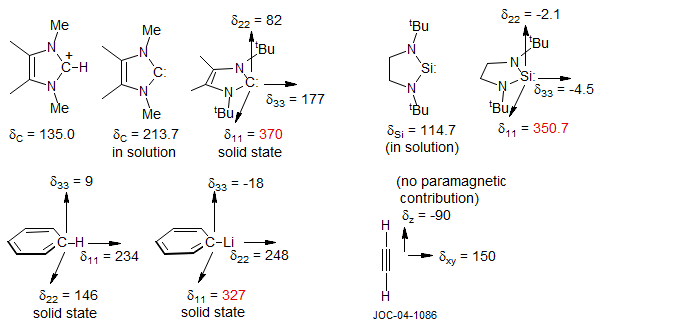
A very similar example of these chemical shift effects is provided by the comparison between methyllithium and phenyllithium. Here the C-Li bond plays the same role as the N-lone pair in pyridine. The conversion of CH4 to CH3Li causes a low-frequency shift, whereas the conversion of C6H5-H to C6H5-Li causes a large high-frequency shift of the ipso carbon.

The individual vector components of the chemical shift can provide additional insights into these effects. The three components of the 13C chemical shift tensors of the carbene below, measured by solid state NMR techniques (Arduengo et. al J. Am. Chem. Soc. 1994, 116, 6361) are shown. The δ11 component which dominates the strong high frequency chemical shift of the carbene carbon (213.7 ppm in solution) is the one perpendicular to the sp2 lone pair on carbon and the empty p orbital, a situation very similar to that in PhLi above. The chemical shifts for all heavy nuclei are dominated by the paramagnetic term. The three components of the 29Si (I = ½) chemical shift of the silylene below (R. West, G. Buffy, M. Haaf, T. Mueller, B. Gehrhus, M. F. Lappert J. Am. Chem. Soc. 1998, 120, 1637) show a similar pattern. The unusual 13C chemical shift of the ipso-carbon in phenyllithium mentioned above also shows similar behavior in the solid state - the δ11 vector component shows a very large downfield contribution, the other two are fairly similar to those in benzene (Berger Chem. Ber. 1995, 128, 1183).
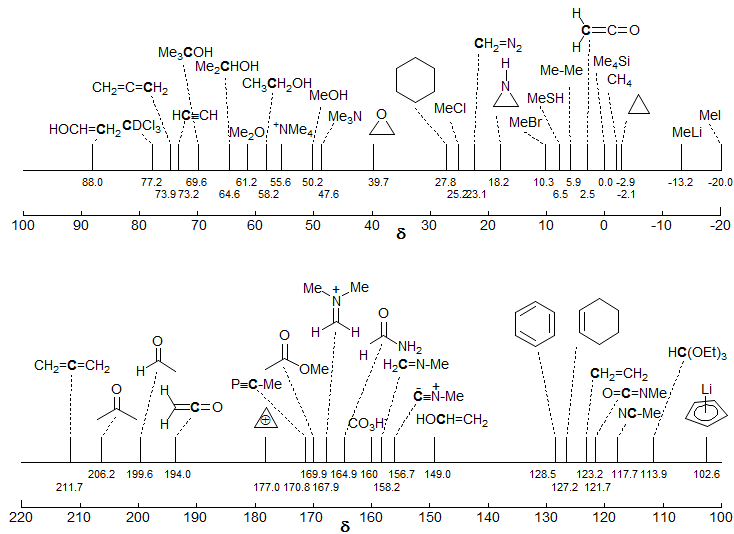
The C-13 Chemical Shift Scale. The vast majority of 13C chemical shifts fall in the range of 0-220 ppm (Me4Si = 0.0). A rough grouping can be made according to the hybridization of the carbon atom, with sp3 carbons at lowest frequency (0-70 ppm); sp carbons of the acetylene type at 70-100 ppm, sp2 carbons (bonded to C and H) at 100-150 ppm, sp2 carbons of carbonyl groups at 160-220 ppm, and sp carbons of the allene type at 210-220 ppm. The chemical shift ranges given in the graph below are not the complete range, since unusual combinations of substituents can lead to shifts outside the ranges given.
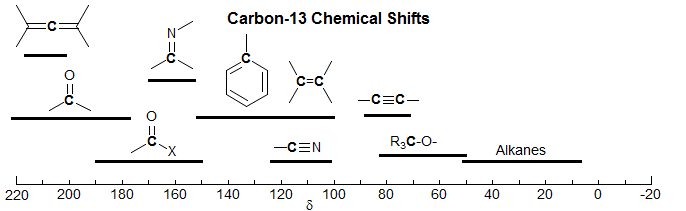
An important point to note is that sp2 carbons of the double bond type cannot be distinguished from those of the aromatic type. This is in contrast to the situation with proton NMR, where the distinction between protons bonded to vinyl carbons and aromatic carbons can often be made easily. Some specific chemical shifts of parent members of the various functional groups are given below. The chemical shifts of more highly substituted compounds will generally be to higher frequency.
Next Section: sp3 Carbons · Previous Section: 13C Spectra ·Home