6-CMR-3 13C Chemical Shift Effects on sp3 Carbons
Changes in 13C chemical shifts are usually discussed in terms of substituent perturbations (Δδ) on the chemical shifts of simpler model compounds. The effects are largest for substituent changes at the carbon itself (α effects) but sizable substituent effects are seen at the β, γ, and sometimes even the δ position. The substituent effects of a number of common functional groups are summarized below. Roughly speaking, the α-effects are strongly dependent on electronegativity of the substituent, the β-effects are almost all to higher frequency, and fairly similar in size, and γ-effects are all to lower frequency (except for organometallic substituents) and are in part the result of steric interactions.
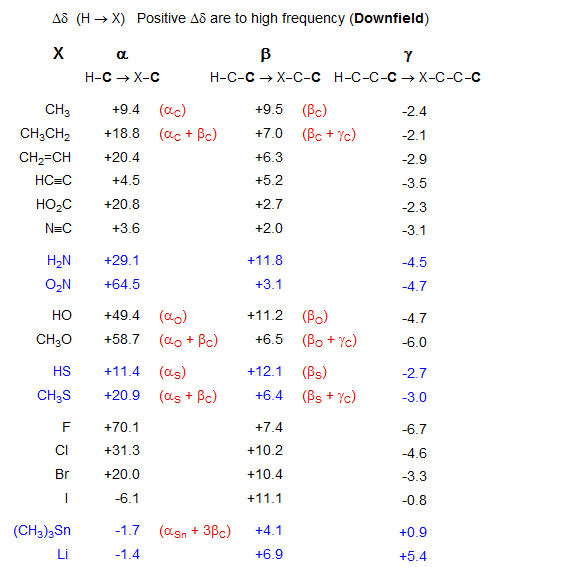
Note: This table is not suitable for detailed chemical shift calculations: use the "n-iso Table" or Grant-Paul parameters for this.
6-CMR-3.1 α-Substituent 13C Chemical Shift Effects
The α-effect results from the replacement of a directly bonded H by an X group (*C-H --> *C-X). The principal factor influencing most α-substituent effects is the electronegativity of the attached atom. Thus, for electronegative atoms we see strong high-frequency shifts (e.g., CH3OH δ 48.8), for electropositive substituents, low-frequency shifts (e.g. Me4Si δ 0.0). For complex groups we must consider β and γ interactions as well (e.g., X = OCH2CH3 is αO + βC + γC). As molecules get more crowded, both the α and β shifts become smaller (branching effects).
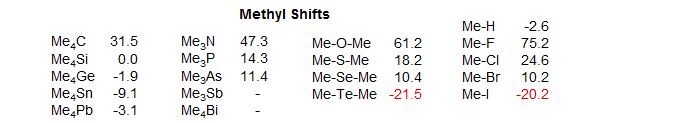
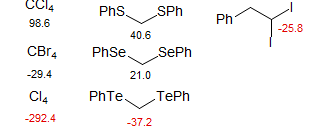
Heavy-Atom α-Effect: The correlation with electronegativity works well for first, and to some extent, second row atoms, but there is a "heavy atom" effect which runs counter to electronegativity. Thus iodine- and tellurium-bearing carbons of all types are strongly shifted to lower frequency (-δ) - note the Me-X and Me-Y-Me shifts above. There is some indication that carbons bonded to Br and Se are also shifted upfield slightly. Heavy atom effects with Ge/Sn and Sb/Bi are less dramatic.
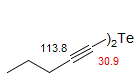
Double Bond α-effects. Unlike the situation with proton NMR, where double bonds cause relatively large downfield (+δ) shifts of allylic protons, the 13C shifts of carbons directly attached to double bonds are changed relatively little compared to a carbon in the analogous saturated alkane. Terminal vinyl groups or trans double bonds cause small downfield shifts, cis-substituted ones cause upfield shifts. The latter effect is a manifestation of the γ-effect.
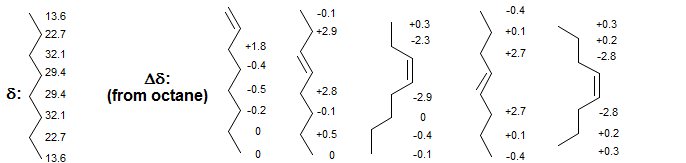
Cycloalkenes sometimes show larger double bond substituent effects, but the size and even direction is erratic, as can be seen from the 3,4,5,6 and 7-membered ring examples below..
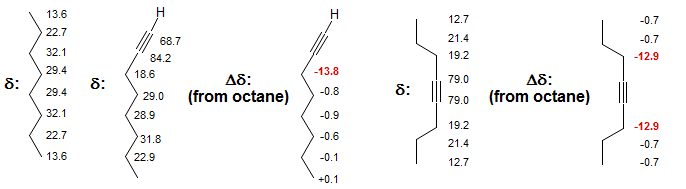
Triple Bonds α-Effects: Triple bonds (X = acetylene, nitrile) as substituents also cause unexpectedly large low-frequency shifts (e.g., CH3-CN δ 0.3, CH3C≡CH at -1.9, compare with CH3-CH=CH2 at 18.7). The large diamagnetic circulation in the triple bond may be in part responsible for these shifts. Below is a comparison of the chemical shifts of octane versus 1-octyne and 4-octyne.
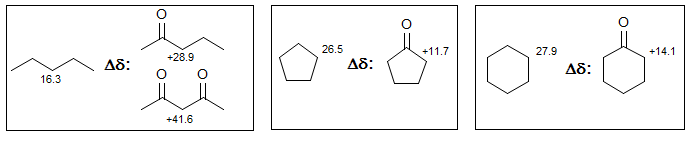
Carbonyl substituents, on the other hand, do cause significant high-frequency shifts.
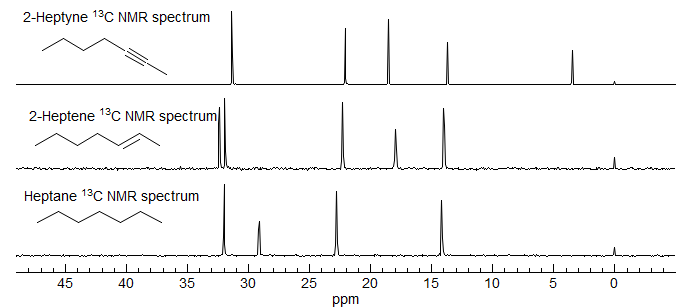
Aside: In contrast to the situation in carbon NMR, where carbons attached to double bonds are barely moved from ordinary sp3 carbons, the downfield chemical shifts caused by double and triple bonds in proton NMR are substantial and generally allow distinction between allylic/propargyllic protons and simple aliphatic ones. In the spectra of heptane, 2-heptene and 2-heptyne below, note that in the 13C NMR the aliphatic carbon shifts span the same range in all three compounds (except for the upfield methyl of heptyne), whereas in the 1H NMR spectra of 2-hexene and 2-hexyne the allylic and aliphatic protons appear in distinct ranges.
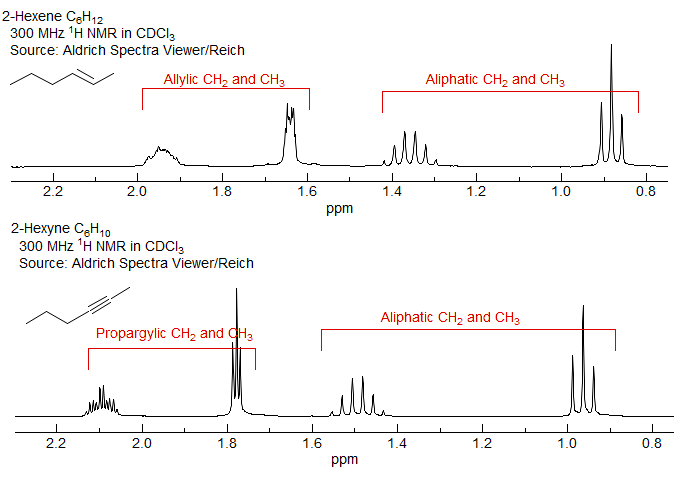
Note: the "ugly" appearance of the two allylic multiplets in 2-hexene is the result of a "virtual coupling" effect: the two vinyl protons are strongly coupled, hence anything coupled to them is messed up.
6-CMR-3.2 β-Substituent Effects
Replacement of H on an adjacent atom by an X group (*C-C-H --> *C-C-X) is a β-substituent effect. Almost all substituents (C or heteroatoms) cause substantial high-frequency (downfield) β-shifts. These are usually around 9-10 ppm, but they are smaller if either the observed or the α-carbon is tertiary or quaternary. Their size is not correlated with the electronegativity of the perturbing substituent. As for the α-effect, we have to consider simultaneous γ-shifts (e.g., Δδβ is smaller for X = CH2-CH3 than for X= CH3, because it is reduced by the negative γC effect, which is absent for Δδβ in X = CH3). The origins of β shifts are not well understood.
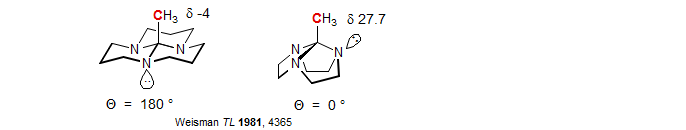
β-Effects of atoms with lone pairs include a stereoelectronic component resulting from electron donation by n-σ* interactions, illustrated by the CH3 chemical shifts below:
6-CMR-3.3 γ-Substituent Effects
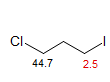
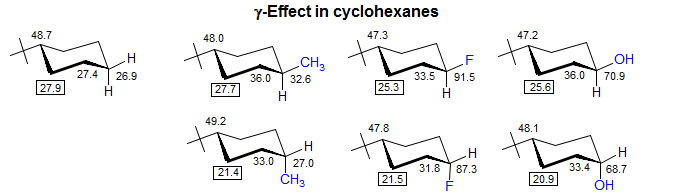
A gamma effect is defined as the replacement of an H by X on the second atom (*C-C-C-H --> *C-C-C-X). Significant γ-effects are seen for virtually all X-substituents, provided the γ-carbon has an attached hydrogen. There is a strong proximity component (syn γ-effect, γ-gauche effect), which results in a dependence on stereochemistry. Syn γ-effects are to low-frequency (Δδ is negative). The effect is largely independent of the nature of the intervening groups. For X = CH3, the effect is upfield by ca 6 ppm if X and *C are close in space (gauche or eclipsed). For acyclic systems, the γC-effect is approximately -2 ppm, reflecting the fraction of the gauche conformation. The γ-effect is extensively used for stereochemical assignments. If a γ-atom is close to a carbon in one isomer, and remote in another, then that carbon will be upfield in the first isomer, as illustrated below (for a theoretical analysis see: Kleinpeter, E.; Seidl, P. R. J. Phys. Org. Chem. 2005, 18, 272). For structure identification using gamma effects see: 1, 2, 3, 4, 5, 6, 7.
The effect is valuable for distinguishing E and Z isomers of alkenes, especially trisubstituted ones, where other techniques (such as 3JHH) are not available. A CH3 (or other carbon group such as a C=C, C≡C, C=O or C≡N) which is cis to a substituent will be to lower frequency (upfield) of the isomer in which the carbon substituent is cis to a hydrogen The effects are very similar across C=N double bonds.
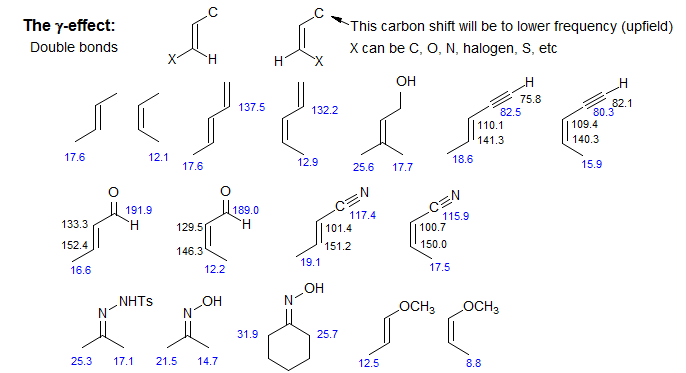
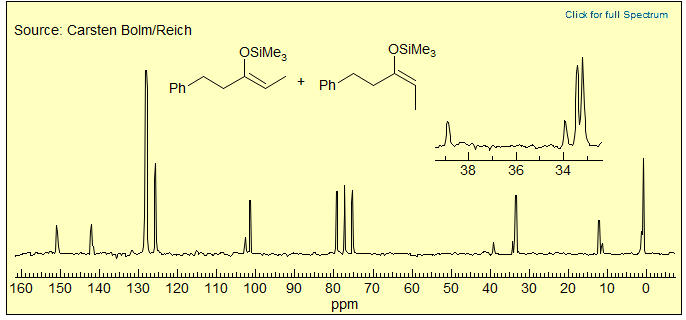
Exercise: The 15.7 MHz 13C NMR spectrum (CDCl3) below is of an approximately 3:1 mixture of stereoisomers. Which is the major isomer? Explain.
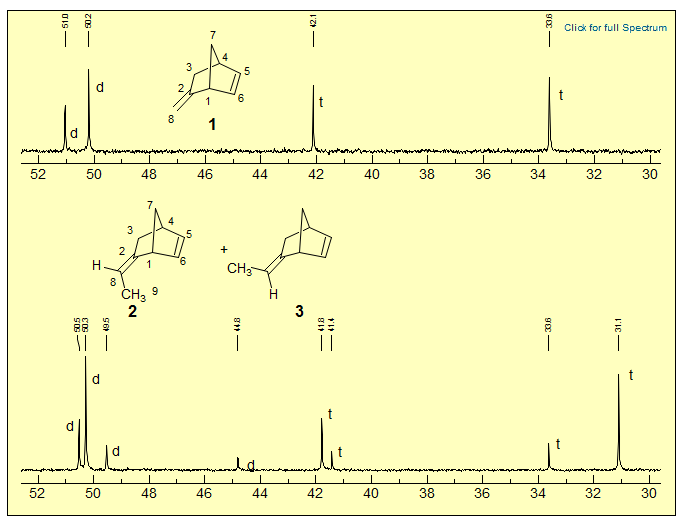
Exercise (R-03E). Assign the aliphatic carbons of 13C NMR spectra of 2-methylenebicyclo[2.2.1]heptene (1) and of a mixture of stereoisomeric 2-ethylidenebicyclo[2.2.1]heptenes (2, 3). Which is the major isomer? Explain.
Stereochemical relationships in a variety of cyclic compounds can be deduced from the presence or absence of γ-gauche interactions. In particular cis-related substituents in cyclopentanes and 5-membered heterocycles cause upfield shifts, not only in the carbons of the substituent, but also in the intervening carbons, compared to trans isomers..
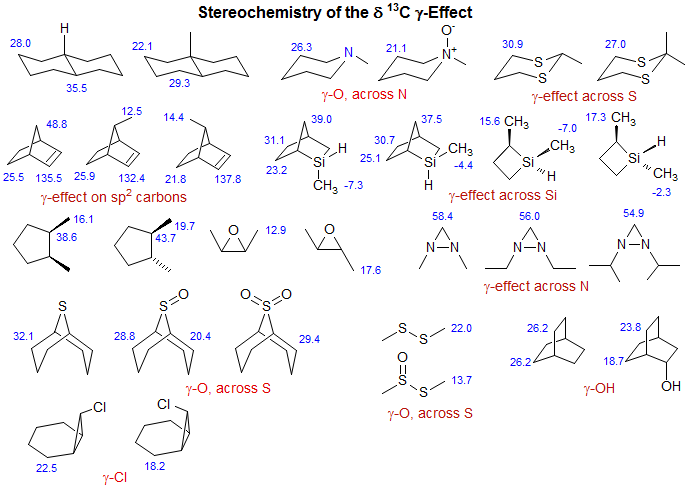
Some substituents also cause anti γ-effects, i.e. when X and the γ-carbon are antiperiplanar. Alkyl substituents show very small antiperiplanar γ-shifts, but for X = O, N, or F significant effects are seen. Anti γ-effects are almost always smaller than gauche γ-effects and they can be either to lower or higher frequency depending on a variety of structural factors. For example, if the perturbing substituent is at a quaternary center in a cyclohexane (as in 1-methyl-1X-cyclohexanes) then the anti γ-effects are to high frequency (downfield), whereas they are to lower frequency (upfield) when the substituent at C-1 is H (Schneider, Hoppen J. Org. Chem. 1978, 43, 3866 DOI).
Determination of Acyclic Syn-Anti Stereochemistry. The γ-interactions present in axial substituents provide the basis for configurational assignment of syn and anti 1,3-diols using the methyl group chemical shifts of their acetonide derivatives. In the syn isomers of the acetonides the 6-membered ring has a well-defined chair conformation, with both R-substituents equatorial. This places one of the acetonide methyl groups axial, the other equatorial, leading to a ca 10 ppm shift difference between the two methyls. The anti acetonides have a twist boat conformation, which places the two methyls in a very similar environment, and hence there is a very small chemical shift differences between them (Rychnovsky Tetrahedron Lett. 1990, 31, 945).

In addition to providing configurational information for systems with well-defined gauche/anti or cis/trans relationships as in the systems above, the generalized upfield shifts of all four of the atoms involved in gauche interactions can also be the basis for stereochemical assignments of diastereomeric pairs in acyclic systems. Thus syn and anti 1,3-diols show a well-defined upfield shift for C-O carbons in the anti compared to the syn isomer. The rationale for this behavior is that intramolecularly H-bonded conformations place a substituent in a pseudo-axial orientation in the anti isomers, hence upfield shifts, whereas all substituents can be equatorial in the syn isomer. Similar shift effects are found in boronic acid esters, where this conformational effect is more rigorously enforced. The effect is easily quantitated by summing the δ values of the two C-O carbons - the one with lower Σ will be the anti isomer (Hoffmann Tetrahedron Lett. 1985, 26, 1643; Chem. Ber. 1985, 218, 3980; for applications see: Pelter Tetrahedron 1993, 49, 3007).
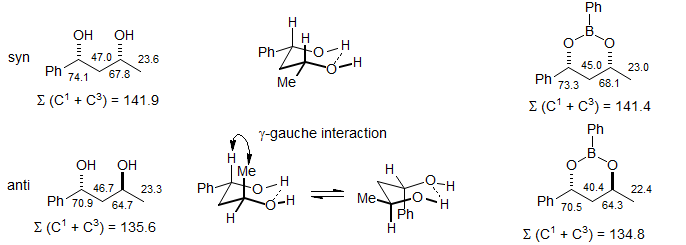
The stereochemistry of aldol adducts (β -hydroxy ketones and esters) can also be determined from 13C chemical shifts by application of similar arguments (Heathcock, Pirrung, Sohn J. Org. Chem. 1979, 44, 4294). The stereochemistry of 1,2-diols can also be determined from analysis of 13C shifts using related arguments.
6-CMR-3.4 δ-Substituent Effects
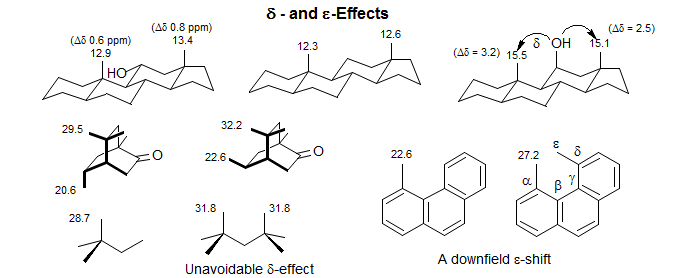
Remote substituents effects across single bonds are small (δ 0.2 ppm, < 0.1 ppm) unless groups are jammed into each other, (e.g., cis 1,3-diaxial) in which case downfield shifts of several ppm are seen (for a theoretical analysis see: Kleinpeter, E.; Seidl, P. R. J. Phys. Org. Chem. 2005, 18, 272).
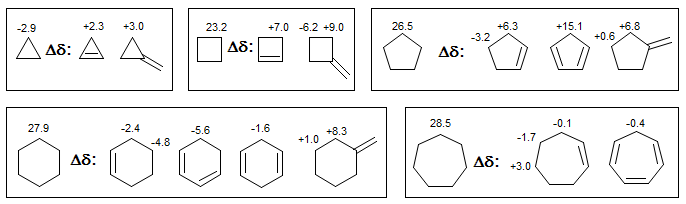
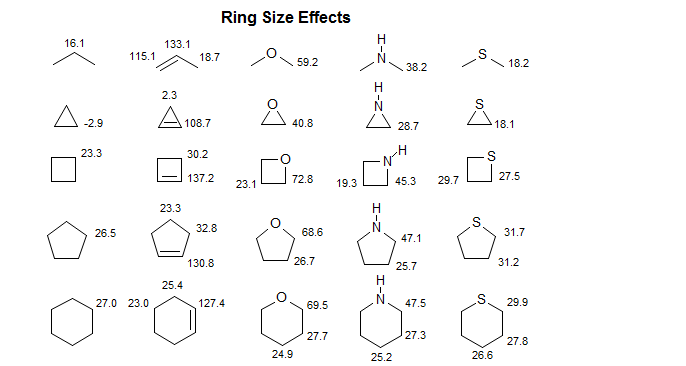
6-CMR-3.5 3-Membered Rings
Cyclopropanes, cyclopropenes, epoxides, aziridines and other 3-membered rings tend to show pronounced upfield shifts. Cyclobutanes and four-membered heterocycles do not show similar effects.
6-CMR-3.6 Neighboring Group Anisotropy Effects
These effects, which play such an important role in 1H NMR spectroscopy, are usually overshadowed by other effects in heavier nuclei. This is because anisotropy effects are the same size (in ppm) for all nuclei. A very striking 2 ppm shift in a proton NMR spectrum will be an (almost) insignificant 2 ppm shift for a carbon at the same position in the molecule (e.g.; it is often trivial to distinguish vinyl from aromatic protons from their chemical shift alone, this distinction cannot be made in the 13C spectrum).
6-CMR-3.7 Alkane 13C Chemical Shift Calculations
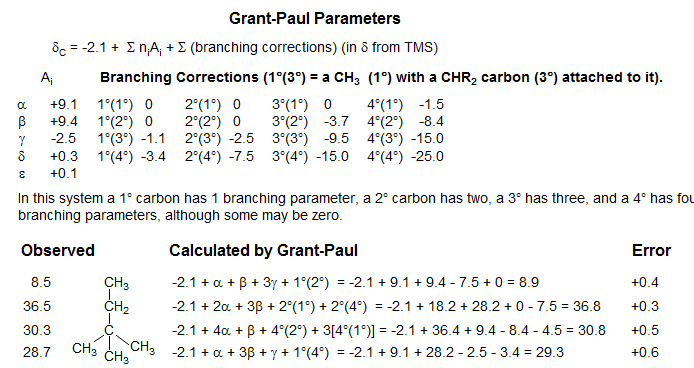
Analysis of the 13C chemical shift of acyclic alkanes led to the first accurate method for the prediction of chemical shifts. Grant-Paul Calculations (J. Am. Chem. Soc. 1964, 86, 2984, plus later improvements) are based on the observation that, in addition to α, β, γ, and δ effects, there are predictable branching effects, such that the α and β, effects, which are nearly constant for linear molecules, become progressively smaller when there are nearby tertiary and quaternary carbons. This is illustrated in the graphic below.
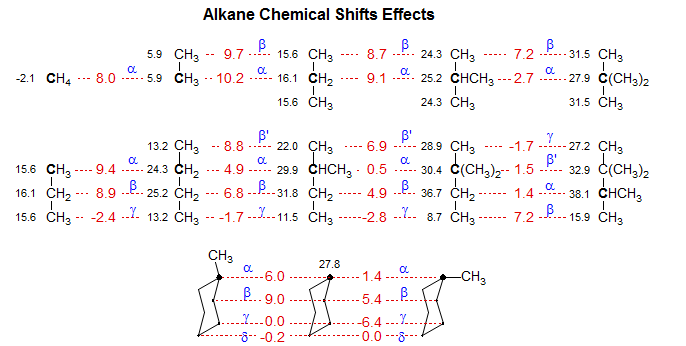
The Grant-Paul system uses a constant set of α parameters, but then applies branching corrections which depend on the number of adjacent branched carbons. The method is quite flexible, and provides a basis for accurately predicting chemical shifts of most alkanes, and can be extended to include other classes of molecules, using model systems close in structure to the molecule of interest.
Substituent Effects Across Heteroatoms. The Grant-Paul β and γ substituent parameters seem to work remarkably well even across heteroatoms like N (in amines) and O (in ethers).

Functionalized Alkane 13C Chemical Shift Calculations
A less extensively parameterized but much more general scheme for the estimation of chemical shifts of alkanes substituted by a variety of functional groups is given below. Instead of branching parameters, this system uses two types of α and β parameters - those for the substituent at the end of the chain (n) and those in which the substituent is in the middle of it (iso). The smaller values of the iso versus the n α and β parameters correspond to the branching corrections of the Grant-Paul system (basically for α the difference between 2°(2°) and 3°(2°), and for β the difference between 2°(2°) and 2°(3°) ). A few parameters have been added for the estimation of shifts for quaternary carbons, but these are not very reliable. Carbons attached to quaternary carbons would require β-quat parameters, and these are not available. One would expect this much simpler system to produce poorer results, and it does. It is permissible to mix Grant-Paul and the n-iso systems in the same calculation, as long as the same effect is not counted twice.
The "n-iso" 13C Parameter Table:
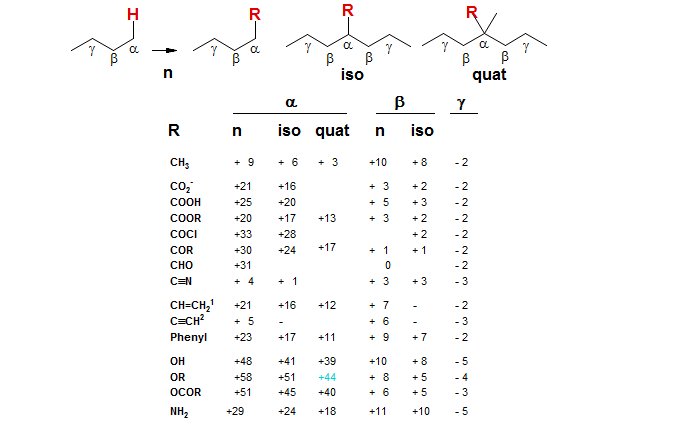
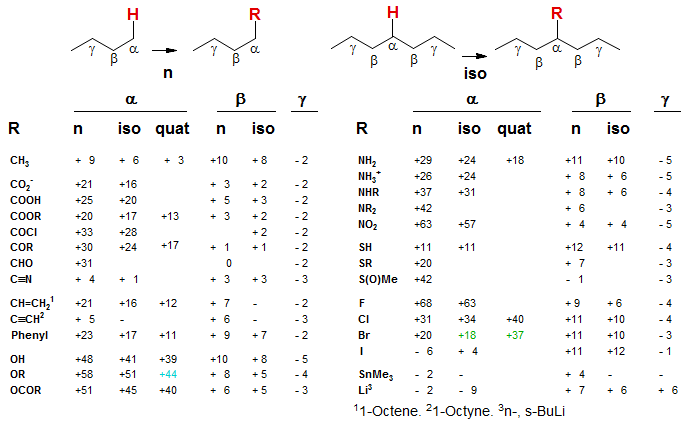
To use this system, the chemical shifts of an appropriate model system are corrected for the presence of substituents by using the parameters in the table. For example, to estimate the C-2 chemical shift of 1-phenyl-2-methylpropane we use the C-2 shift of isobutane (23.3) and add the β-Ph-n increment (+9), giving 32.3 (observed 30.1). Similarly, C-1 would use C-1 of isobutane (24.6) and α-Ph-n increment (+23) giving 47.6 (obs 45.3). In this case the shifts are estimated with reasonable accuracy.
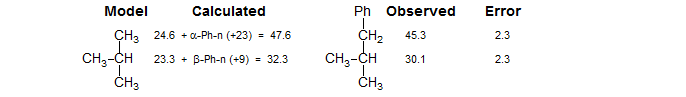
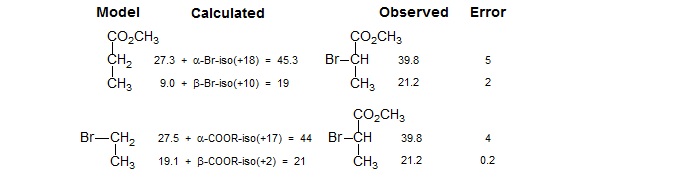
However, attempts to use this method to calculate the C-2 shift of methyl 2-bromopropionate are a little less successful. Using methyl propionate as a model, C-2 is in error by 5 ppm, whereas using bromoethane as model the error is 4 ppm. It turns out that the shift parameters for Br are quite sensitive to the local environment, and the result is larger errors in shift estimates.
13C Chemical Shift Calculations
1. For acyclic alkanes the Grant-Paul Parameters are the most effective. They can be used either to calculate shifts completely (from methane), or by a difference method (perturbation). Branching parameters can get complicated during such calculations.
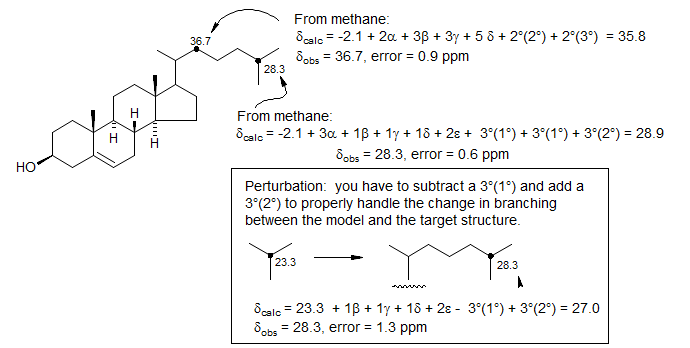
2. For more complicated systems, use model compounds as close as possible to the actual structures, and then apply corrections for any structural differences using Grant-Paul and other parameters (including branching corrections). If needed, a chemical shift parameter can be calculated by comparing two model compounds.
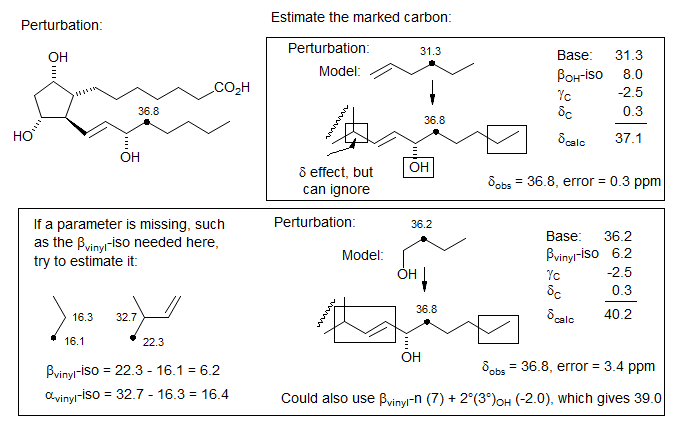
Another example of mixing the Grant-Paul and the n-iso parameters is shown below. The error in the calculated shift is in the direction expected for an underestimation of the upfield branching effects of the adjacent quaternary carbon. A guess at this correction would be the difference between 2°(4°) (-7.5) and 3°(4°) (-15), i.e -7.5. Applying this correction gives an error of only 2.2 ppm.

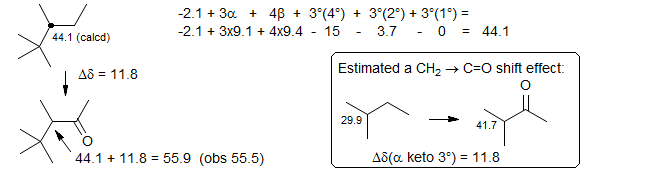
We could also use an approach to the above calculation which takes the branching more specifically into account - estimate the hydrocarbon using Grant-Paul parameters, and add a "customized" α-keto effect (replacing a CH2 by a keto), which we calculate ourselves from some model systems:
Next Section: sp2Carbons · Previous Section: Carbon Shifts·Home