6-CMR-5 One-Bond Carbon-Proton Coupling (1JCH)
Coupling between protons and carbon was first observed in 1H NMR spectra when 13C satellites were detected. All C-H signals are flanked by 0.55% 13C satellites more or less symmetrically disposed around the C-H signal (there is a small isotope shift, so the satellites are not exactly centered on the central peak corresponding to the 12C-H signal). These result from the one-bond coupling between 13C and 1H, and are separated from each other by 100-300 Hz (i.e., separated from the central peak by 50-150 Hz). The 1JCH values of most simple molecules were measured from analysis of the 13C satellites. An example (N-methylmaleimide) is shown below. The 13C satellites of the N-CH3 group are singlets, separated by 140.3 Hz. Interestingly, the satellites of the vinyl protons (1JCH = 184.2 Hz) are doublets, because the isotopic label now makes the two protons non-equivalent, and we can detect the coupling between them (6.0 Hz).
It is also possible to observe satellites from 2JCH (HCC) and 3JCH (HCCC) coupling. Since these signals will be only a few Hz from the central peak which is 200x as large, careful tuning of the spectrometer, and a relatively simple 1H spectrum (preferably sharp singlets or doublets) are required before such satellites can be detected.
More commonly, 13C NMR spectra without decoupling (coupled spectra) are used, especially for the measurement of longer-range couplings. These spectra are often rather complex, and if the 1H spectra are second order (strongly coupled) or form magnetically non-equivalent spin systems (e.g., AA'BB') then the coupled 13C NMR spectra will also be second order (e.g., the X part of an AA'BB'X system), and computer simulations of the carbon spectra (and sometimes also the proton spectra) may be required to extract the various nJCH couplings.
For a tabulation of carbon-proton couplings see J(C-H) data.
The size of 1JCH is correlated closely with the hybridization of the C-H bonding orbital - the values are roughly proportional to the % s-character. This arises because the principal coupling mechanism is the Fermi contact term, which involves transmission of coupling information between the nuclei via the s-electrons (only s-electrons have finite electron density at the nucleus).
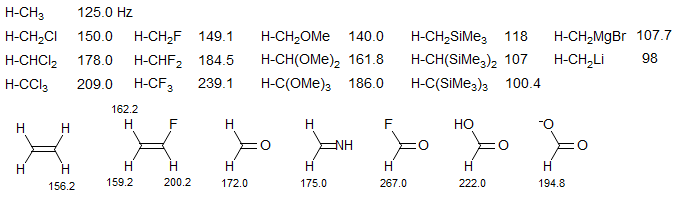
The above formulas can be used to estimate hybridization for simple hydrocarbons, but the method works less well for systems having electronegative substituents. For these the coupling constants go up consistent with increased s-character in the C-H bond (see CHF3) but the molecular structures do not show the large distortions from tetrahedral geometry that would be expected from simple application of these formulas.
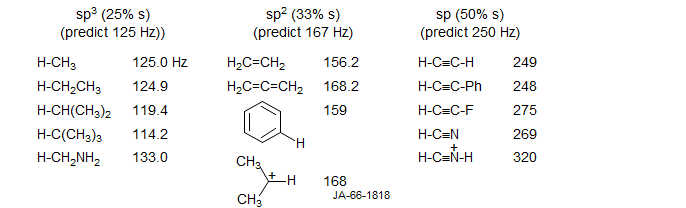
Electronegative substituents cause increases in 1JCH. electropositive substituents a decrease.
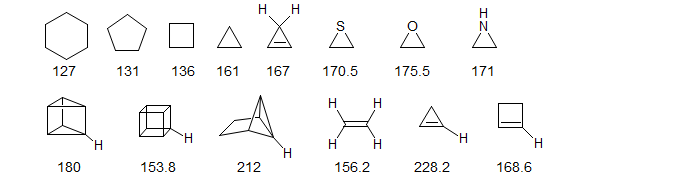
Strained rings show unusually large C-H couplings, consistent with the idea that carbons in such rings have high s-character in the C-H bonds and high p-character in the endocyclic C-C bonds.
Hybridization effects on 1JC-H in Carbanions
Organometallic compounds of electropositive metals generally show the effects of significant rehybridization to accommodate the negative charge on carbon. For localized lithium and magnesium reagents there is a tendency for the "lone pair" orbital to maximize s-character, resulting in enhanced p-character for the C-H bonding orbitals. This can be seen in the sequence of methyl derivatives, where the 1JC-H becomes progressively smaller as the substituent becomes more electropositive. Similarly for the vinyl series.
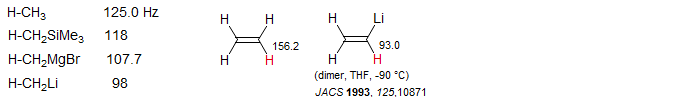
For lithium reagents where π or σ delocalization is possible, the opposite effect can be seen, in that the coupling becomes larger as the C-M bond becomes more polarized, or is broken entirely. This is assigned to increased planarization of the carbanion center (towards sp2 hybridization) to maximize π overlap. Both effects can be seen for benzyllithium - in benzene solution the benzyl carbon is strongly coordinated to lithium (in an aggregate) and J is smaller than for the methyl CH in toluene, whereas in THF, and even more so for benzylpotassium the J becomes larger as the carbanion becomes less strongly coordinated by the metal cation. The chemical shift behavior can also be interpreted in these terms, with downfield shifts corresponding to increased planarization (Waack, McKeever, Doran Chem. Commun. 1969, 117; Bauer J. Am. Chem. Soc. 1994, 116, 528)
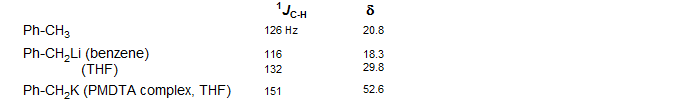
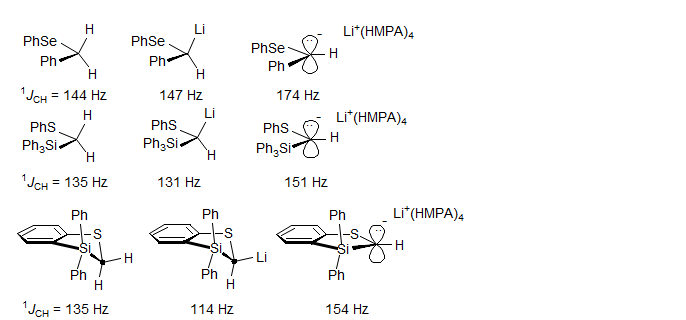
The effect of ion pair separation for lithium reagents which are delocalized is generally to increase the iJC-H as a result of planarization of the carbanion center, as seen in the examples below (Reich, H. J.; Dykstra, R. R. J. Am. Chem. Soc. 1993, 115, 7041).
Stereoelectronic Effects on 1JC-H
There are well-defined stereoelectronic effects on the C-H couplings of axial and equatorial protons in cyclohexanes (Perlin effects). These are most pronounced for situations where strong electronic interactions of n→σ* type or σ→π* type cause perturbations of the C-H bond length and bond strength either by electron donation into the C-H σ* orbital or electron withdrawal from the σ orbital. In cyclohexane itself the axial C-H coupling is slightly smaller than the equatorial (ΔJ = 4.0 Hz), attributable to a stronger donation by the axial H-C σ bond, compared to the equatorial C-C bond. In 1,3-dioxane the difference is much larger (ΔJ = 10.1 Hz), reflecting the larger donation by the p lone pair on each oxygen.
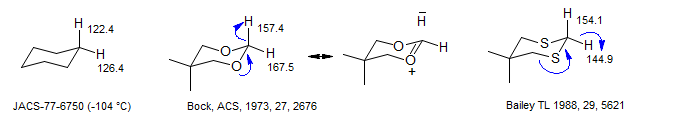
It is interesting that dithiane shows an inverse effect, attributed to a relatively weak n→σ*CH interaction, and a stronger σCS→σ*CH interaction. There are other indications that n→σ*CH interaction in dithianes is weak. For example, the conformational equilibrium of H vs D (measured by the Saunders' isotope perturbation method) indicates no preference for 4,4-dimethyl-1,3-dithiane, but a significant preference for C-H to be axial for the 1,3 dioxane and diazane (Anet Chem. Comm. 1987, 595).
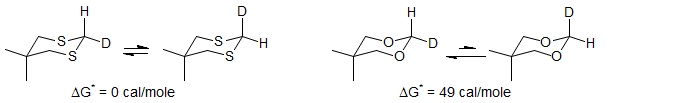
Next Section: 2JC-H · Previous Section: Shifts vinyl ·Home