7-MULTI-2 Effects of Other NMR-Active Nuclei on 1H and 13C NMR Spectra.
There are many NMR active nuclei other than 13C and 1H that are encountered by chemists. The effects on proton and carbon NMR spectra can vary from negligible to dramatic, depending on the natural abundance of the X-nucleus, and on its nuclear properties (spin and T1 relaxation).
7-MULTI-2.1 J Coupling with 100% Abundant Nuclei Spin = ½ Nuclei (19F, 31P, 89Y, 103Rh)
For the 100% abundance nuclei 31P and 19F nearby protons and carbons will be split due to JHX and JCX coupling. The interpretation of such spectra can be difficult, since splitting due to chemical shifts and couplings cannot be directly distinguished. There are several techniques for making such distinctions, including decoupling experiments (e.g., 1H 31P or 13C 31P), which may require special spectrometer accessories (triple-resonance probes), or measurement of spectra at different field strength, where peak separations due to couplings will be invariant in Hz, whereas those due to chemical shifts will be invariant in ppm. In the two 13C NMR spectra in Fig. 7.2.1 there were several ambiguities and one superimposed peak which were resolved in this way. For other spectra involving coupling to P see: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20. For spectra involving coupling to F see: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.
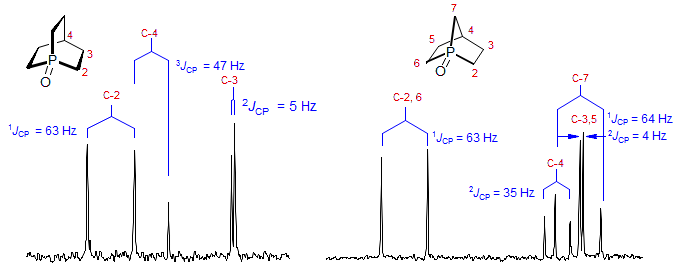
Figure 7.2.1. 25 MHz 13C NMR spectra of two bicyclic phosphine oxides (Wetzel, Kenyon J. Am. Chem. Soc. 1974, 96, 5189).
Exercise: Analyze the partial 13C 1H NMR spectrum of the phosphonate below. Estimate J-values.
Exercise: Identify all of the 1H-31P couplings in the spectrum below.

Exercise: Here is a more complicated system, where all of NMR-active nuclei in the ring are coupled to each other. Analyze the 1H NMR multiplets and Identify all of the 1H-1H and 1H-31P J couplings in the spectrum.

Exercise: Identify the H-H and F-H couplings in 2,4-dinitrobenzene.

7-2.2 Low Abundance Spin ½ Nuclei (Si, Sn, Pb, Se, Te, Fe, Pt, Xe, N, Hg, etc)
Spin ½ Isotope Satellites. For less abundant X nuclei, satellites on the main 1H peak will be observed from J-coupling. The intensity of each satellite will be half of the abundance of that isotope. For examples see: Si: 1, 2, 3; Sn:1, 2, 3, 4, 5; Pb: 1; Se: 1, 2, 3, 4, 5, 6, 7, 8; Te: 1; N-15 1 Xe: 1.
When there are several X-nuclei of this type coupled to a C or H, one does not see multiplets (triplets, quartets) since the large majority of molecules have only one active nucleus. Rather, the size of the satellites increases in proportion to the number of substituents (e.g., the 1H signal of H2-C(SePh)2 will have 77Se satellites that are twice as high as those of CH3-SePh). However, coupling to multiple low-abundance nuclei will not be completely invisible, the satellites will themselves have satellites due to presence of doubly-labelled molecules (the tiny fraction of H-C(77SePh)2 present - (0.07)2 = 0.56%). For examples see: 1, 2.
The spectrum below shows a mixture of two selenium compounds, PhSe-CH2-Br and PhSe-CH2-SePh. The satellite intensities allow a decision on which CH2 peak is which - the smaller upfield peak at δ 4.22 has 77Se satellites that are, relatively, twice as large as those on the downfield CH2 peak at δ 4.72. Thus the assignment has to be as shown.

Some elements (e.g. Sn, Te) have more than one NMR active spin 1/2 nucleus, and so more than one set of satellites can be observed. For these the size of the coupling constants will be in the ratio of the gyromagnetic ratios, and the intensities will be in the ratio of the natural abundances of the isotopes. An example is shown below in the 1H NMR spectrum of PhTe-CH2-TePh. For the central CH2 peak the satellites for 125Te and 123Te can be detected, and even the 125Te satellites on the 13C satellite are visible (green expansion).

Tin is even richer in spin 1/2 isotopes. There are three, but only two are present in enough abundance to appear in normal spectra. In the spectra below the satellites of the two most abundant isotopes can be seen, as can 1H isotope shifts due to deuteration.

Figure 7.2.2. 500 MHz 1H NMR spectra of a tin compound, illustrating satellites and isotope shifts. The outer satellites therefore correspond to 119Sn, the isotope with the largets gyromagnetic ratio and abundance. Satellites due to 115Sn can barely be detected in this spectrum. The spectra also show how replacement of a CH3 by a CD3 group can be followed by proton NMR, using the isotope shift of CD3 on the geminal CH3 group -- the exo-CH3 is replaced by CD3 much faster than the endo-CH3. (Reich, H. J.; Borst, J. P.; Coplien, M. B.; Phillips, N. H. J. Am. Chem. Soc. 1992, 114, 6577).
7-2.3 Size of Coupling Constants to Spin 1/2 Nuclei
Although a detailed discussion of coupling between H and the numerous other NMR-active nuclei is (currently) beyond the scope of these notes, there are some generalizations that are useful: The size of coupling between protons and some commonly observed spin 1/2 nuclei like 31P, 19F, 15N, 29Si, 119Sn, 77Se, 125Te, 199Hg follows the same trends as do H-H couplings in comparable environments
1. Because of more effective orbital overlap, the trans 3J coupling across double bonds is always larger (by a factor of two or so) than the cis coupling. Note the highly variable size of the two-bond 2JH-X coupling










2. For similar reasons, 3-bond couplings across single bonds follow Karplus-like rules. Thus the anti-periplanar 3-bond coupling is typically larger than gauche or syn periplanar. This can be seen in the JH-F and the JH-Hg values below. Examples: 1, 2


Exercise: Analyze the 1H NMR spectrum of benzylselenol below, and extract the 1H - 77Se J couplings.

Exercise: Determine all available couplings (JHH, JHPb, JCPb), and measure the chemical shift difference between the CH3 and CH2 protons in tetraethyllead (the CH3 and CH2 protons of the A3B2 system are too close together to resolve in the central peak).

Exercise: Assign the protons and determine all available couplings (JHH, JHSn) in the vinyl group of tributylvinyltin.

Exercise: Three-bond coupling constants across double bonds for most nuclei follow the same rules as do H-H and C-H couplings: Jtrans is almost always greater than Jcis. Assign the two vinyl protons in the compound below using this information. Click spectrum for answer.

7-2.4 Second Order Effects
The rules for second order effects are the same as for proton NMR spectra: if two nuclei which are coupled to each other are also very close in chemical shift, then any nucleus coupled to these will show second order effects. An example is provided by the behavior of phosphine and phosphite complexes of metals. If a complex has two such ligands, and they are equivalent by symmetry, they form an AA' system, and, provided that there is a significant coupling constant between them, all nuclei coupled to the P atoms will show virtual coupling effects. If JPP is large, all nuclei coupled to them will be triplets, or more complex patterns, even though such nuclei may be coupled to only one of the P atoms. An example is provided by the carbon NMR spectra of lithium diphenylphosphide in Figure 7.2.3:

Figure 7.2.3. 13C NMR spectra of lithium diphenylphosphide dimer in ether. At low temperature the ipso and ortho carbon signals are triplets. Because the two phosphorus nuclei are coupled to each other (2JPP must be greater than 80 Hz to explain the spectra) the carbons are the X part of an ABX (CPP') system with a very small ΔδAB (the 12C/13C isotope shift on the P nuclei which are the AB part), and thus the carbons appear to be coupled to both P nuclei (virtual coupling). At higher temperature the DNMR exchange of Li-PPh2 units becomes fast on the NMR time scale (probably by dissociation to monomeric LiPPh2 units), the P-P coupling is lost, and a normal doublet is seen for the 13C signals (Reich, H. J.; Dykstra,, R. R. Organometallics 1994, 13, 4578).
This effect is frequently seen in transition metal P-ligand complexes where phosphine or phosphite ligands are symmetry related. All proton and carbon signals coupled to one of the P nuclei become triplets, or more complicated patterns, depending on the size of 2JPP. Examples: 1.

One application of this effect is as an aid in determining the stereochemistry of octahedral and square planar transition metal complexes. Since trans phosphine ligands typically show much larger 2JPP values than do cis ligands, trans phosphines often show apparent triplets for P-Me groups, whereas cis phosphines show normal doublets or more complex intermediate signals (this is not the case, however, in the example above, where both cis and trans phosphite pairs show full virtual coupling).
Another example is provided by the C-4 signal in 1,3-difluorobenzene (Fig. 7.2.5). Here the two fluorines are coupled to each other and have only a very small chemical shift difference (due to 12C/13C isotope shifts, estimated at 1.25 Hz at 56.4 MHz), and thus the carbon signals will be second order. the (X part of a strongly coupled ABX (FF'C) system.

Figure 7.2.5. 13C NMR signal of C-4 of 1,3-difluorobenzene (Weigert, F. J.; Roberts, J. D. J. Am. Chem. Soc. 1971, 93, 2361).
7-MULTI-2.5 J Coupling with Quadrupolar nuclei (Spin > ½)
In principle, a neighboring quadrupolar nucleus should split a proton or carbon signal into a multiplet of the 1:1:1 .. type (1:1:1 for I = 1, 1:1:1:1: for I = 3/2, etc) instead of the 1:1 doublet seen for spin 1/2 nuclei. This is because such nuclei have more than two possible orientations in the magnetic field (for I = 1, m = 1, 0, -1). In practice, such coupling is seen only for a few select cases, because for most such nuclei quadrupolar T1 relaxation is too fast to allow detection of the coupling. For a neighboring quadrupolar nucleus X the effect depends principally on the spin (I) of X, the symmetry of the molecule, and the electric quadrupole moment Q. T1 of the X nucleus will be short if the quadrupole moment is large, and the nucleus is not at the center of tetrahedral or octahedral symmetry. In this case the nucleus may be effectively "self decoupled." This is why we see little or no effects on nearby H and C nuclei (other than small chemical shifts) of 35Cl (I = 3/2, Q = 0.05), 79Br (I = 3/2, Q = 0.3), and 127I (I = 5/2, Q = 0.7). At intermediate T1 values of X, partial averaging of C-X or C-H J coupling can result in line broadening. This is often the case for 14N (I=1, Q = 0.016) and 11B (I = 3/2, Q = 0.036). For example, some 14N-H protons are very broad as a result of this effect (this is actually a T2 effect on H). If X has a low quadrupole moment, or is at a center of high symmetry, then T1 of X can be long, and multiplets are observed for the spin ½ nucleus. Thus 1:1:1 triplets are often observed for nuclei coupled to 2H (I = 1, Q = 0.0028) and 6Li (I = 1, Q = 0.00046). See summary

An example of coupling between 19F and 133Cs in an encapsulating molecule.

7-2.6 Deuteration
Substitution of 1H by 2H (D, I = 1,Q = 0.00287) is an important tool in organic chemistry, and is probably the most frequently used isotopic substitution. Deuterium is commonly used as a mechanistic probe, and deuterium incorporation can often be easily done, e.g. by base catalyzed exchange of acidic CH protons. The NMR properties are quite good, quadrupolar relaxation is relatively slow so that lines are reasonably sharp and the very small couplings to deuterium (1/6 the size of coupling to protons) can sometimes be resolved. For Examples of H-D, C-D and F-D coupling see: 1 (H), 2 (H), 3 (H), 4 (F), 5 (C), 6 (C), 7 (H).
Effects of Deuteration on 1H NMR: When a proton is replaced by deuterium, there are three changes in the 1H NMR spectrum - loss of signal, changes in coupling and isotope shifts. The most obvious effect is that the corresponding signal disappears in the 1H NMR spectrum. This effect is commonly used to detect protons at exchangable sites (O-H, N-H, S-H) where the signal can be identified by simply adding D2O to the sample and observing changes in the NMR spectrum (loss of signal and loss of coupling, if any).

The second effect is changes in coupling: coupling constants of different isotopes are in proportion to their gyromagnetic ratios. Thus JHD will be ca 1/6 as large as JHH (γD / γH = 1/6.488). For many situations the coupling will effectively disappear, leaving only some line broadening (a typical 7 Hz 3JHH will become a 1 Hz JHD). There might also be a broadening effect resulting from the slightly shorter T1 of D versus H. For larger coupling constants the doublet splitting of e.g. 17 Hz for a trans 3JHH coupling across a double bond will be replaced by a ca 2.7 Hz 1:1:1 triplet for 3JHD, as in the example below, which also illustrates the loss of coupling for the terminal vinyl protons (1.5 Hz 2JHH becomes 0.2 Hz 2JHD).

For an example of deuterium isotope shifts on a proton NMR spectrum see Section 5-HMR-3.9.
Effects of Deuteration on 13C NMR Spectra: The effects of replacing hydrogen by deuterium in 13C NMR spectra are as follows:
1. The CD, CD2, or CD3 signal can almost disappears in the 13C NMR, although the resulting D-coupled carbon signal can be detected at high signal to noise. The large loss in intensity is the result of three factors: (i) splitting the peak into a multiplet (1:1:1 triplet for CD, 1:2:3:2:1 quintet for CD2, 1:3:6:7:6:3:1 septet for CD3, 1JCD = 20-30 Hz), (ii) loss of signal due to saturation (dipole relaxation becomes very inefficient – the carbon has the NMR properties of a quaternary carbon), and (iii) reduction in NOE enhancement (Section 8-02).
2. The deuterium-coupled multiplet will be upfield from the protio analog by about 0.3 ppm (isotope shift - see below).
3. Carbons that are one and two bonds removed from deuterated carbon (or O-D, N-D, S-D etc) will show reduced peak height since there will be small residual two-bond and three-bond couplings to the deuterium (2JCD, 3JCD is typically 1-2 Hz) causing the peak to become broader (see Fig. 7.2.7). Under ideal conditions 2JCD and 3JCD can be resolved. There will also be small isotope shifts, usually upfield. (see Fig. 7.2.8 and Fig. 7.2.11).
An example of this technique that was used to aid in the assignment of the 13C chemical shifts of the steroids is shown in Fig. 7.2.6, and of a steroid analog in Fig. 7.2.7.

Figure 7.2.6. 15 MHz 13C NMR spectra of androstane-3,17-dione and a deuterated analog obtained by base-catalyzed H-D exchange. Note that signals for the deuterated carbons C-2, C-4 and C-16 have disappeared, and that those for some neighboring carbons are slightly reduced in intensity (C-1, C-5, C-10, C-14, C-15). These are CW spectra taken on one of the first NMR spectrometers capable of taking high-resolution 13C NMR spectra (Reich, H. J.; Roberts, J. D.; Weigert, F. J. J. Am. Chem. Soc. 1969, 91, 7445).

Figure 7.2.7. 25.2 MHz 13C NMR spectrum of homo-androst-3.6-dione in CDCl3. The lower spectrum is of a sample treated with NaOCD3/DOCD3. Note that the signals for C-2, C-4 and C-7 are nearly gone, that of C-5 is greatly reduced (partial deuteration here). The signals of those carbons that are 2 or 3 bonds from a D show broadening and hence reduced peak height (e.g. C-1, C-8, C-9, C-10, C-14) (from Abraham, Loftus).
7-2.7 Effect of Boron on NMR Spectra
Boron has two NMR-active isotopes, 10B (19%, Q = 0.086 I = 3) and 11B (81%, Q = 0.036, I = 3/2). The relatively favorable Q value and high abundance means that 11B NMR is very useful in investigating boron compounds. Nuclei such as 1H and 13C coupled to boron (as well as the boron resonances themselves) tend to be broad, and sometimes difficult to resolve because of the relatively short T1 values caused by quadrupolar relaxation. A spectrum of the BH protons in a phosphine-borane is shown below. The 1JH-B is ca 95 Hz. Examples: 1, 2, 3.

The NMR signals of carbons bonded to boron are broad, and sometimes difficult to observe. Coupling to boron is usually not resolved.

Figure 7.2.8 below shows an example of a 10B/11B isotope shift observed in the 19F NMR spectrum of tetrabutylammonium tetrafluoroborate. The two resonances are separated by 0.05 ppm with an intensity ratio of approximately 20:80 corresponding to the natural abundances of 10B and 11B. The low frequency resonance is due to 11BF4-. Since 11B is a spin I = 3/2 nuclide we observe a 1:1:1:1 quartet with J = 2 Hz corresponding to the one bond 19F - 11B coupling. The high frequency resonance is due to 10BF4-. Since 10B is a spin I = 3 nuclide we expect a very poorly resolved 1:1:1:1:1:1:1 septet with J = 0.4 Hz corresponding to the one bond 19F - 10B coupling.

Figure 7.2.8: 19F NMR spectrum of tetrabutylammonium fluoroborate Source.
7-2.8 Effect of Nitrogen on NMR Spectra
Nitrogen has two NMR-active isotopes, 15N (0.39%, I = 1/2) and 14N (99.64%, Q = 0.019 I = 1). Both nuclei have low gyromagnetic ratios (frequencies of 10 and 7 MHz with 1H = 100). This has several consequences - direct observation of each nucleus usually requires a special probe, the signals are relatively insensitive, and coupling constants to both isotopes of nitrogen are small. The relatively short T1 values caused by quadrupolar relaxation ("self-decoupling") of the 14N in most compounds means that fully resolved coupling to the major isotope 14N is only seen in rare instances where the nitrogen has high symmetry, such as in symmetrical quaternary ammonium salts. In fact, in the vast majority of nitrogen compounds, there is little indication of broadening of C-N carbon signals, or H-C-N proton signals due to residual coupling to 14N. However, the N-H protons of amides (see formamide, other amides: 1, 2, 3, 4, 5) often show substantial broading due to residual effects of partially relaxed coupling (there may also be broadening due to dynamic proton exchange).
It is possible, in simple compounds, to detect 15N satellites in proton NMR spectra (they are 0.2% the intensity of the central peak). In complex molecules the small size of the J values and the low intensity of the satellites makes their observation very challenging. Thus, most information about 15N shifts and coupling constants comes from isotopically enriched materials (15NH4Cl is relatively inexpensive).
7-MULTI-2.9. Isotope chemical shifts
NMR chemical shifts are detectably altered by close proximity to isotopes of different mass, and such isotope shifts are easily detected on high field spectrometers for the lighter nuclei (where the ratio of masses of pairs of isotopes is relatively high). Such shifts have turned out to be very useful for determining the presence and amounts of isotopic labels. For examples, see Fig. 7.2.2 above for a remote deuterium isotope shift on a proton signal, which allows accurate determination of deuterium incorporation from the 1H NMR spectrum, Fig. 7.2.8 above for an isotope shift on 19F caused by 10B/11B (fluorine is especially sensitive). In Sect. 7-MULTI-3 there are additional examples such shifts: Fig 7.3.3 (D on 119Sn), Fig. 7.3.4 (18O on 31P) and Fig 7.3.5 (D on 77Se).
See Fig. 7.2.9 and 7.2.10 below for the effect of deuterium on 13C NMR spectra. Other examples of isotope shifts: 1, 2, 3.

Figure 7.2.9. 100 MHz 13C NMR spectra of cyclopentane, cyclohexane, norbornane and their mono-deuterated analogs with 1H broadband decoupling in CDCl3 (Aydin, Günther J. Am. Chem. Soc. 1981, 103, 1301). Estimate the isotope shifts in ppb (parts per billion = 1000 ppm) for the various carbons.

Figure 7.2.10. 25.2 MHz 13C NMR spectrum (noise decoupled) of a 1:1 mixture of 2-methylcyclohexanone and its 2,6,6-trideutero analog (inserts are five-fold expansions). The best way to estimate D incorporation at C-2 is to compare the areas of the two methyl carbons at δ 15 (Wehrli79 p.108).
Problem R-98G: Analyze and identify all peaks in the13C NMR spectrum of a mixture of CH2Cl2, CDHCl2 and CD2Cl2. .

Figure 7.2.11. 100 MHz 13C NMR spectrum of a mixture of CH2Cl2, CDHCl2 and CD2Cl2. The spectrum is NOT proton decoupled.
Next Section: 7.3 - Spin 1/2 Nuclei · Previous Section: 7.1 - Nuclear properties · Home