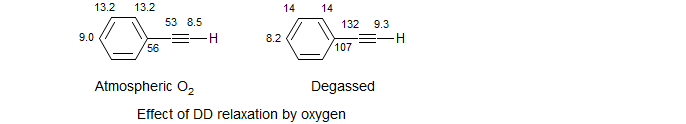8-TECH-1 Relaxation in NMR Spectroscopy
An understanding of relaxation processes is important for the proper measurement and interpretation of NMR spectra. There are three important considerations.
1. The very small energy difference between α and β states of a nuclear spin orientation in a magnetic field results in a very small excess population of nuclei in the ground vs the excited states (typically less than 1 out of about 10,000 molecules). For many nuclei relaxation (i.e., return from excited to ground state) is a very slow process, with half-lives on the order of 0.1 to 100 seconds for a spin ½ nucleus (compare this with micro-, pico- and femtoseconds for relaxation of electronic and vibrational transitions). It is thus very easy to saturate an NMR transition (equalize populations of excited and ground state), with the resultant loss in signal quality, and failure to obtain correct peak areas.
2. NMR lines are extraordinarily sharp, and extraordinarily close together (in energetic terms) compared to higher energy spectroscopic methods. So much so that Heisenberg uncertainty broadening (which is a function of lifetime of a given energy state, and hence relaxation rates) is a dominant feature of many NMR spectra, and can limit our ability to measure and interpret spectra. When relaxation is very fast, NMR lines are broad, J-coupling may not be resolved or the signal may even be difficult or impossible to detect.
3. The success of many multipulse experiments, especially 2D and 3D spectra, depends crucially on proper consideration of relaxation times.
T1 and T2 Relaxation. We distinguish two types of relaxation, Spin-Lattice (T1, also known as longitudinal relaxation, or relaxation in the z-direction) and Spin-Spin (T2, also known as transverse relaxation, or relaxation in the x-y plane). T1 relaxation corresponds to the process of establishing (or re-establishing) the normal Gaussian population distribution of α and β spin states in the magnetic field. T2 is loss of phase coherence among nuclei. T2 is less than or equal to T1 (R = relaxation rate, R2 = 1/T2, R2 ≥ R1), since return of magnetization to the z-direction inherently causes loss of magnetization in the x-y plane. The line width of an NMR signal is determined by T2 - short T2 means broader lines (ν1/2 = 1/πT2, ν1/2 = width at half height). The maximum repetition rate during acquisition of an NMR signal is governed by T1 - short T1 means the magnetization recovers more rapidly, and a spectrum can be acquired in less time.
8-TECH-1.1 Sources of Line Broadening in NMR - T1 and T2 Relaxation
1. Instrumental problems - tuning, mis-shapen NMR tube, etc
2. Sample problems:
· Sample inhomogeneity (poor mixing. solid particles)
· Temperature gradients across sample
· Paramagnetic impurities
3. T1 Relaxation (Spin-Lattice Relaxation): gain and loss of magnetization in the z-direction. NMR lines are at least as wide as specified by the Heisenberg Uncertainty Principle broadening due to inherent lifetime of spin states (the actual width is governed by T2). For most spin 1/2 nuclei T2 is between 0.2 and 50 seconds.
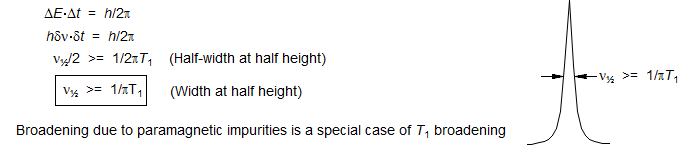
Place unmagnetized sample in magnetic field: establishment of the normal Boltzmann equilibrium between the α and β spin states has a first order rate constant of k = 1/T1: Mz = M∞ (1 - e-t/T1)
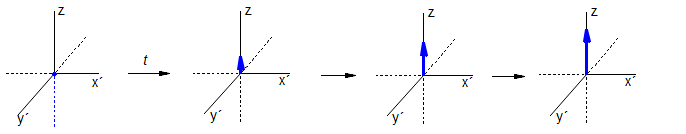
Or apply a 90° pulse, and watch the magnetization return to the z-direction: Mz = M∞ (1 - e-t/T1)
The rate of spontaneous relaxation of nuclear spin orientations is almost zero. T1 relaxation is caused by transient magnetic fields (usually due to molecular motion) at the Larmour precession frequency. In most situations T1 relaxation is optimal if the average rate of molecular reorientation in space is at the Larmour precession frequency. In mobile liquids near room temperature the average rates of molecular rotation are several orders of magnitude higher than ν0, so only a very small fraction of the motions are at the proper frequency, leading to very inefficient relaxation (long T1). For larger molecules and more viscous solutions molecular motions become slower and and more efficient relaxation results (T1 shortens). However, at some point the average molecular motions become slower than ν0, and T1 becomes longer again (Figure 8.1-1).
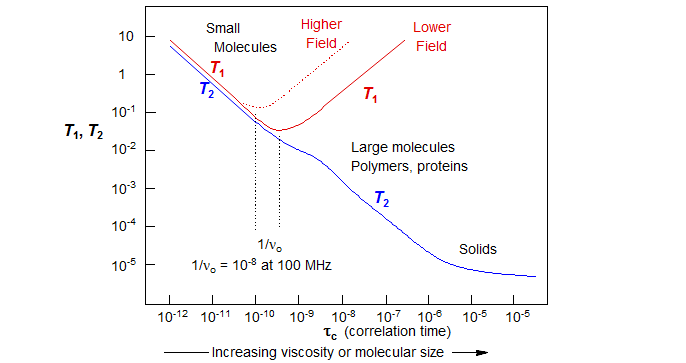
Figure 8.1-1. Behavior of T1 and T2 as a function of correlation time for spin ½ nuclei relaxing by the Dipole-Dipole mechanism. τ = Molecular correlation time: the time it takes the average molecule to rotate one radian (adapted from Bloembergen, E.M. Purcell, R.V. Pound "Relaxation Effects in Nuclear Magnetic Resonance Absorption" Physical Review 1948, 73, 679-746)
4. T2 Relaxation (Spin-Spin Relaxation): Heisenberg Uncertainty Principle broadening due to lifetime of spin coherence - gain and loss of magnetization in the x,y-direction.
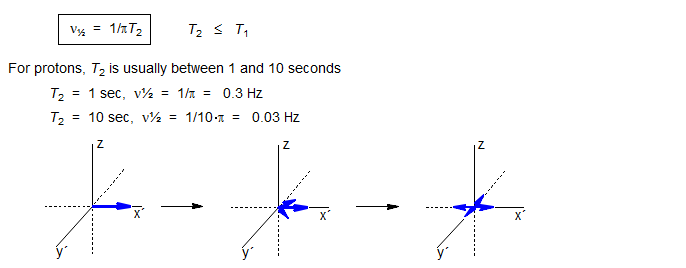
T2 relaxation is caused by transient magnetic fields (usually due to molecular motion) at any frequency. Thus T2 keeps getting shorter as molecular reorientation rates slow down (Figure 8.1-1).
T2 relaxation is also caused by swapping of chemical shifts or coupling constants due to chemical exchange or interconversion of conformations. Line broadening due to chemical exchange provides an important tool for measurement of the rates of a variety of molecular processes (see Sect 8-TECH-3).
A third source of T2 relaxation is interconversion of spin states of a quadrupolar nucleus coupled to it
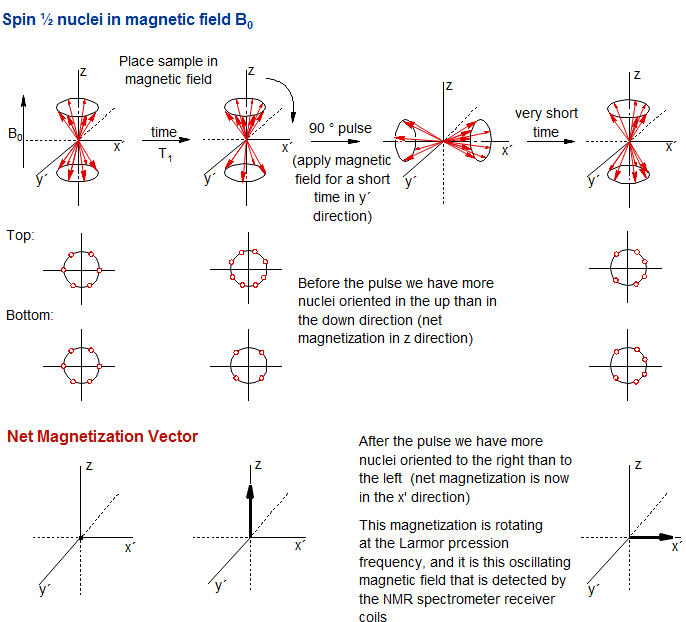
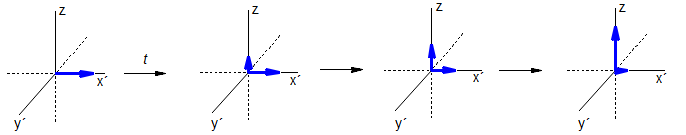
Figure 8.1-2. Symbolic presentation of nuclear spins in response to a 90° pulse. The population difference between the α and β spin states is greatly exaggerated
Measurement of T1 by Inversion-Recovery
Measurement of T1. A common method is the 5T1-π-τ-π/2-FID pulse sequence. A series of NMR spectra are measured in which the spins are inverted with a π pulse, followed by a variable waiting period τ (Figure 8.1-3). The degree to which the signal has returned to its equilibrium value during the waiting period τ for each sequence is measured, and plotted as a first order rate process. A sample set of spectra for such an experiment is given in Figure 8.1-4.
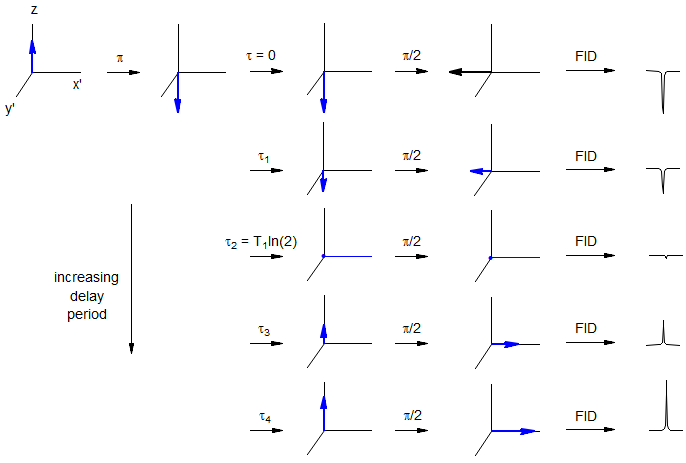
Figure 8.1-3. Pulse sequence for a T1 determination by the inversion-recovery method.
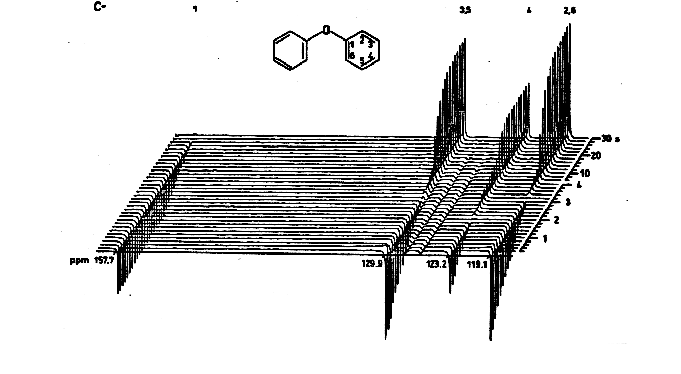
Figure 8.1-4. Example of an inversion-recovery T1 determination. From "13C NMR Spectroscopy, High Resolution Methods and Applications in Organic Chemistry and Biochemistry," E. Breitmaier and W. Voelter, Verlag Chemie, Weinheim, 1987, page 52.
8-TECH-1.2 Relaxation Mechanisms
Spontaneous T1 relaxation of spin ½ nuclei, i.e., relaxation in the absence of external influences, is essentially absent. For T1 relaxation to occur there must be magnetic field fluctuations in the x,y direction. Such fluctuations are most effective when they occur at the Larmor precession frequency (νo). T1 relaxation is thus field dependent, since νo varies with the field.
T2 relaxation is caused by fluctuations in any direction.
The principal source of fluctuating magnetic fields in most molecules is molecular motion. We can define a correlation time τc for a molecule (assuming it behaves more or less spherically). This is the average time it takes the molecule to rotate through one radian. The correlation time for small molecules is of the order of 10-12 sec in solution (longer in viscous solvents). Since for a proton at 300 MHz νo ≈ 108, most molecules below molecular weight of 1000 are moving too rapidly for effective relaxation (Figure 8.1-1). There are several mechanisms by which molecular motions can influence nuclear relaxation: direct interactions with nearby magnetic nuclei (DD), chemical shift effects (CSA), quadrupole-electric field gradient interaction (QR) and rapid modulation of J-coupling (SC). In addition to molecular motion, rotational transitions can also be the source of fluctuating magnetic fields (SR).
Below are summarized the pricipal relaxation mechanisms (more or less in inverse order of importance).
8-TECH-1.3 Relaxation by Spin Rotation (SR)
A local magnetic field is generated by the circular motion of electrons in a rapidly rotating molecule (or part of a molecule, such as a methyl group).
The magnitude of this field changes when the rotational energy levels change as a result of molecular collisions. These changes, if they occur at the Larmor precession frequency, can cause relaxation of nearby nuclei. The correlation time for SR relaxation is not molecular motion, but the lifetime of rotational quantum states.
Characteristic features:
· Small molecules, or freely spinning portions of larger molecules without other efficient relaxation mechanisms.
· Better relaxation (shorter T1) at higher temperatures.
· The SR mechanism has been detected for methyl groups, and for quaternary carbons of small molecules which do not have other effective relaxation mechanisms (e.g. CS2). It is also seen for 29Si and 119Sn in small molecules such as Me4Si and Me4Sn as well as mobile end groups such as Me3Si-O-R which do not have directly attached protons to cause efficient DD relaxation or structural features that cause CSA relaxation.
· Operation of this mechanism causes reduction or loss of NOE effects.
8-TECH-1.4 Relaxation by Chemical Shift Anisotropy (CSA)
The chemical shift of a nucleus is a function of the orientation of the molecule in the magnetic field (i.e. it is a tensor quantity), provided the nucleus is not at the center of tetrahedral or octahedral symmetry. For ethylene (13C) acetylene (13C) and nitrobenzene (15N), the limiting chemical shifts are given below:
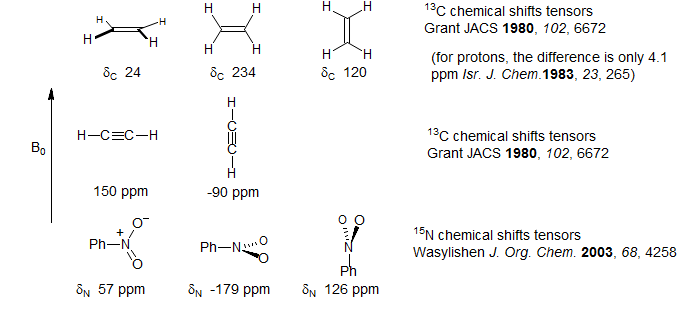
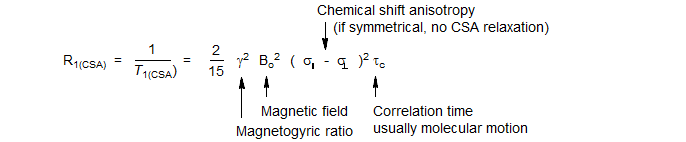
As the molecule tumbles in solution, the chemical shift (and hence the magnetic field at the nucleus) is constantly changing, since the paramagnetic term of the shielding equation is directionally dependent. Thus tumbling can cause relaxation of the nucleus if it happens at the right frequency. The relaxation rate is proportional to the square of the gyromagnetic ratio and of the magnetic field strength, as well to the chemical shift anisotropy. The correlation time is molecular motion. The equation below applies to cylindrically symmetric molecules (one symmetry element):
Characteristic features:
(1) Operation of this mechanism causes reduction or complete loss of NOE effects when protons are decoupled.
(2) The mechanism is never seen for protons (chemical shift anisotropy is too small), and is seen for carbon only when there are no attached protons (e.g., carbonyl compounds other than aldehydes and formates, quaternary sp3, sp2 and sp carbons). On the other hand nuclei such as 199Hg, 119Sn, 195Pt, 77Se, 205Tl, 207Pb and 125Te with large chemical shift ranges are often relaxed largely or entirely by the CSA mechanism, especially at high fields.
(3) The rate of relaxation is proportional to the square of the field strength, and thus becomes much more important at higher field. For some spin ½ nuclei with large chemical shift ranges, lines become sufficiently broadened by CSA relaxation to cause loss of coupling information. For such nuclei, modern high-field spectrometers can be a decided disadvantage. For example, the T1 of Hg(CN)2 goes from 0.18 sec at a field strength of 5.9 Tesla, to 0.063 sec at 9.4 T (Wasylishen Can. J. Chem. 1982, 60, 2113).
This effect is illustrated by the 1H NMR spectra below. Shown is the H2 proton of a platinum complex at 90, 250 and 400 MHz. The pattern consists of the central ddd corresponding to all of the molecules with NMR-inactive Pt nuclei. The pair of satellites are due to the molecules containing the spin 1/2 nucleus 195Pt. Note that at a spectrometer frequency of 90 MHz the satellite signals are nearly as sharp as the central peaks. As the spectrometer field strength (B0) is increased the satellites become broader and are barely recognizable at 400 MHz. At still higher field strength they would begin to merge with the central peaks. These changes are the result of increasing rates of T1 relaxation of 195Pt by the CSA mechanism, which varies as Bo2.
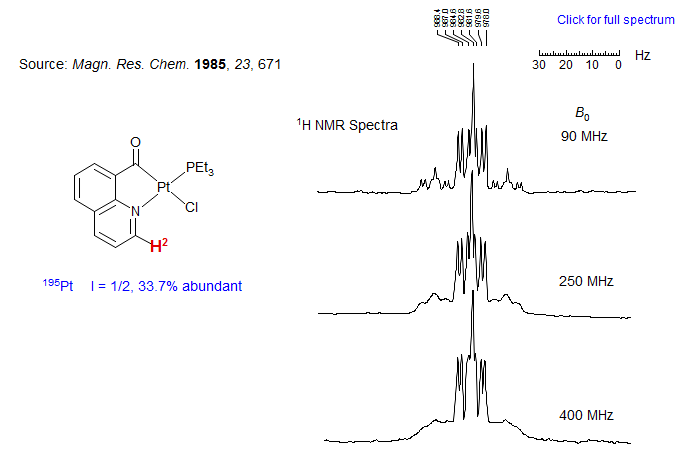
Exercise: Extract and assign all of the coupling constants of the H2 proton.
(4) CSA relaxation is generally dominant in nuclei with large chemical shift ranges, and in bonding situations which produce large magnetic anisotropies. Molecules in which the nucleus is at the center of tetrahedral, octahedral, or higher symmetry (119Sn in Me4Sn. 29Si in Me4Si) have no chemical shift anisotropy and so do not relax by this mechanism. This is illustrated by comparison of the 199Hg T1 of the linear structure Hg(CN)2 and the tetrahedral Hg(CN)42- anion (Wasylishen Can. J. Chem. 1982, 60, 2113).
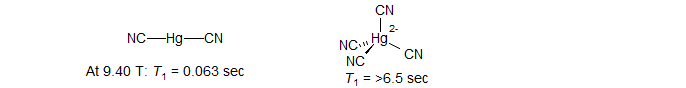
8-TECH-1.5 Quadrupolar Relaxation (QR)
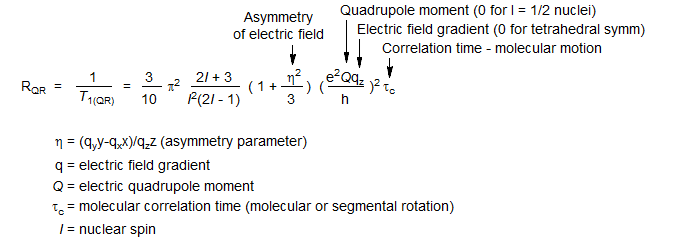
This mechanism operates for spin >½ nuclei only, and works well only for nuclei which are not at the center of tetrahedral or octahedral symmetry. Spin ½ nuclei can be considered to have spherical charge distributions, but for spin >½ nuclei the charge distribution has the shape of an oblate or prolate spheroid. Electric field gradients in such molecules exert a torque on the quadrupolar nuclei. Tumbling of the molecule can then initiate transitions among the spin states - there is "friction" between the nucleus and the surrounding electrons, the "quadrupole coupling constant" e2Qqz/h. The effectiveness of this relaxation mechanism is critically dependent on Q. If Q is small (as for 2H and 6Li, both with I = 1) the behaviour is almost like that of a spin ½ nucleus (i.e. there is only a small deviation from spherical symmetry), if it is large the coupling between the electric field and nuclear assymmetry is strong and the nucleus can have very short T1. Observation can be very difficult, and spin-spin splitting to the nucleus cannot be observed.
In molecules with tetrahedral or octahedral symmetry the electric field gradient is small or zero, and T1 values can be long enough to obtain acceptable spectra (e.g., ClO4- for 35Cl, SO4-2 or R2SO2 for 33S).
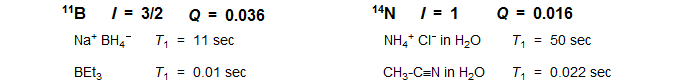
However when Q is large enough, even nuclei in a formally symmetric environment can show fast QR. For example the spherically symmetrical iodide ion (I = 5/2) has such a large quadrupole moment (Q = 0.79) that line widths of from 1200 to >50,000 Hz are seen in various solvents due to transient asymmetric solvation.
Quadrupolar relaxation of a nucleus can also have effects on nearby magnetic nuclei, since rapid relaxation can either broaden or entirely remove J-coupling between the two nuclei. This effect is especially common for 14N-X and 11B-X groups. Such broadening is actually a T2 effect at the X nucleus (interchange of spin states). It is often difficult to observe carbons or protons directly bonded to boron because of this kind of T2 broadening. Rapid quadrupolar relaxation of Cl, Br and I nuclei is the reason we don't normally see any indication of coupling between these nuclei and adjacent carbons or protons (QR is so fast that these nuclei are self-decoupled).
Characteristic features:
· Only for nuclei with I > 1/2.
· Faster relaxation at lower temperatures in solution (better match of νo and motion).
· There must be a permanent (or transient) electric field gradient across the nucleus (i.e., QR is much less effective in molecules in which the nucleus is at a center of tetrahedral or octahedral symmetry).
· For nuclei with a large electric quadrupole moment, this mechanism is so effective that only kHz wide lines can be observed, and no J coupling to other nuclei can be detected.
· For most quadrupolar nuclei no NOE effects are seen when protons are decoupled.
The 7Li and 6Li NMR spectra below illustrate the importance of the quadrupolar coupling Q and the symmetry of the environment around lithium. The 7Li NMR spectra show a relatively sharp peak for the product mercury ate complex (width 4 Hz), where the lithium is tetrahedrally solvated by THF, but a much broader peak for PhLi dimer (25 Hz), and still broader for PhLi monomer (60 Hz), where the environment is less symmetrical than for the dimer. The Q value for 6Li is much smaller than for 7Li, and for 6Li both the monomer (3.5 Hz) and dimer (2.2 Hz) peaks are much sharper. A small part of the broadening of the 6Li spectra may be the result of some dynamic exchange between the monomer and dimer structures, this might explain the greater linewidth of monomer over dimer (Reich, Green, Medina, Goldenberg, Gudmundsson, Dykstra, Phillips, J. Am. Chem. Soc. B, 120, 7201 (Reich, Green, Medina, Goldenberg, Gudmundsson, Dykstra, Phillips, J. Am. Chem. Soc. 1998, 120, 7201; Reich, Green, Phillips, J. Am. Chem. Soc. 1991, 113, 1414). Line width were estimated by simulation with WINDNMR.
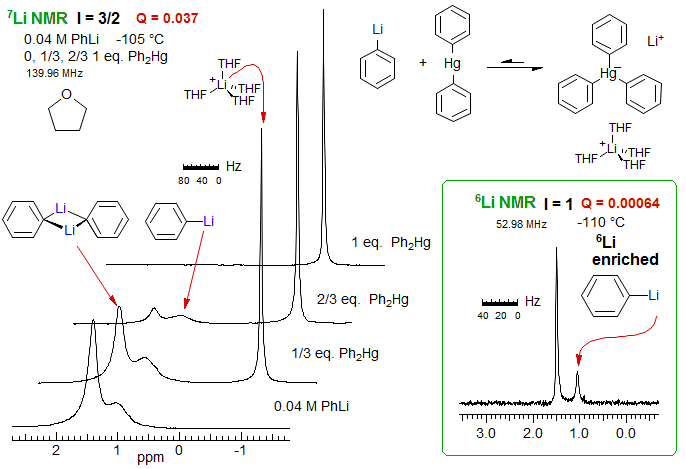
8-TECH-1.6 Scalar Relaxation (SC)
Scalar (J) coupling of a nucleus Y to a second quadrupolar nucleus X can provide a relaxation mechanism for Y if X is undergoing very rapid T1 relaxation. Under these conditions Y is subject to a fluctuating magnetic field because of the rapid spin reorientation of X. For this mechanism to be very effective, the Larmor precession frequencies of X and Y must be very close together (i.e., ωX - ωY must be small).
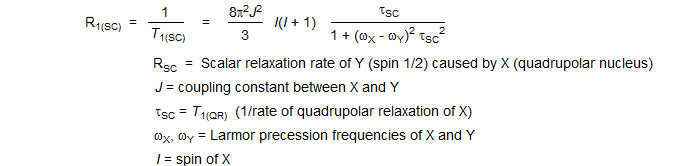
Because of the stringent requirement that the Larmor precession frequencies of the two nuclei involved be close together, this effect is very rare. The unusually short T1 values for bromine-bearing carbons are believed to be caused by SC relaxation. On a 200 MHz spectrometer, the Larmor precession frequency of 13C is 50.28 MHz, that of 79Br (I = 3/2), 50.18 MHz.
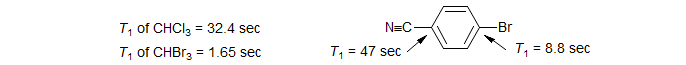
Note that the SC T1 relaxation process is quite distinct from the broadening of Y caused by partially coalesced J coupling between Y and X that occurs when T1(X) has values in the range of 1/(πJC-X) (as is often seen for spin ½ nuclei attached to 14N and 11B). Such broadening is a T2 process in Y (swapping of spin states).
8-TECH-1.7 Dipole-Dipole Relaxation (DD)
Coupling between magnetic nuclei is of two types: the scalar (J) coupling is the result of polarization of the electrons by the nuclear spins. The direct dipole-dipole coupling(D) interaction is very large (often kilohertz) and depends principally on the distance between nuclei and the angular relationship between the magnetic field and the internuclear vectors. This coupling is not seen in mobile solutions because it is averaged to zero by tumbling of the molecule. However, as the molecule tumbles in solution the dipole-dipole coupling is constantly changing as the vector relationships change. This creates a fluctuating magnetic field at each nucleus. To the extent that these fluctuations occur at the Larmor precession frequency, they can cause nuclear relaxation of nearby nuclei. Since the proton has the highest magnetic dipole of common nuclei, it is the most effective nucleus for causing DD relaxation. DD relaxation is the principal relaxation pathway for protons in molecules containing contiguous protons, and for carbons with directly attached protons.
The correlation time for DD relaxation is molecular (or, more accurately, segmental) motion. The equation governing DD relaxation of an X nucleus by nearby protons in the region of fast molecular motion (extreme narrowing region):
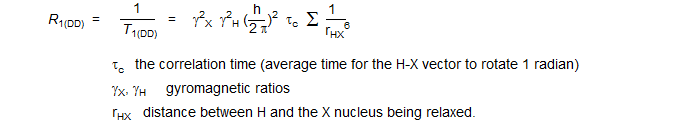
DD relaxation shows a very strong distance dependence, and operates most effectively between directly bonded nuclei. At the usual spectrometer frequencies, small molecules (MW <1000) are tumbling too fast for the most effective relaxation (τc is too short). Thus the more rapidly a molecule or part of a molecule tumbles, the less effective DD relaxation is, and the longer T1 becomes. Large molecules (e.g. proteins) are usually moving too slowly (τc is too long), and they have the opposite relationship between molecular motion and T1 (i.e., relaxation is more effective when the molecule moves faster).
For 13C NMR, dipole relaxation by directly attached protons (if any are present) is the principal relaxation mechanism. Thus quaternary carbons will have long relaxation times (hence their low intensity under normal conditions of spectrum acquisition due to saturation). CH groups will have shorter values, and CH2 groups still shorter by approximately a factor of two. This can be seen in the 13C T1 values of adamantane, and the carbons in the ring framework of cholesteryl chloride. The side chain and methyl carbons have separate (faster) motions, and thus have longer T1 values.
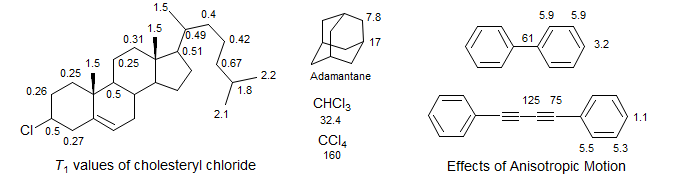
Anisotropic Motion: Long thin molecules do not move isotropically in solution (i.e., τc will be different for rotation around different axes of the molecule), so that relaxation times can be quite different for carbons which are on the long axis, compared to those off it. A nice example is diphenyldiacetylene. The para carbons have relatively short relaxation time (1.1 s) whereas for the ortho and meta carbons T1 is about five times as long. This is because rotation around the long axis is very fast, and causes inefficient relaxation for the off-axis carbons. The para carbons change their relationship with the attached hydrogen only when rotating around an axis perpendicular to the acetylenes, a much slower process. This effect can sometimes be used to assign carbon signals. Note the unusually long T1 of the central acetylene carbons - the result of being very far from any protons.
Segmental Motion: Different parts of molecules can have different correlation times, and consequently T1 values. For 1-bromodecane, the T1 values are uniform down the chain except near the ends, where local conformational motions are somewhat faster. On the other hand, in 1-decanol the T1 values become progressively shorter closer to the OH group, probably a consequence of "anchoring" of the OH end by intermolecular hydrogen bonding. You can also see the effects of segmental motion in the chlolesteryl chloride T1 values - all of ring CH are ca 0.5 sec, the CH2 0.25 sec - that part of the molecule moves as a unit. However, in the side chain the T1 values become longer, up to 0.67 sec for the last CH2 and 1.8 sec for the last CH, because the side chain has additional flexibility, and thus a shorter correlation time.
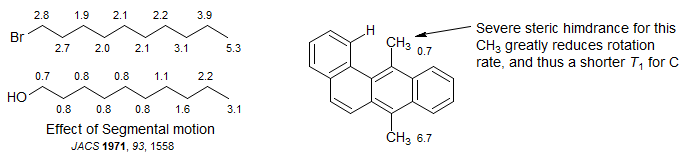
Methyl groups show highly variable T1 values. There are now three directly attached protons and thus one would expect 1/3 the T1 compared to CH carbons. However, unless the CH3 group is very sterically hindered the local motions are much more rapid than the rest of the molecule, and thus the carbon undergoes less effective DD relaxation. This may be in part offset by SR relaxation, which becomes more effective at higher rates of rotation.
Relaxation by Oxygen. The unpaired spins on O2 make it capable of causing proton and carbon relaxation by a dipolar interaction. The effect is not large, but can be easily seen for the quaternary carbons of phenylacetylene - e.g. the ipso carbon T1 increases from 56 to 107 sec if the dissolved oxygen is removed. This can be done by vaccum techniques, or more simply by bubbling N2 or argon through the sample for a minute or two. Normally O2 makes a contribution at the level of T1 ca 50 to 100 sec. Since most T1 values are less than 20 sec, O2 does not normally have much of an effect. Only very long T1 values (>>20 sec) are significantly affected by oxygen.
It is important to remove dissolved oxygen from samples for NMR experiments where long T1 values are important (e.g. in multipulse/2D experiments where long delay times or used), or where proton-proton DD relaxation must be maximized (e.g. in the NOE experiment discussed in Section 8.2).