8-TECH-5 The Saunders Equilibrium Isotope Perturbation Experiment
Consider the question of whether a system with two limiting structures is equilibrating or delocalized (two energy minima or one). Benzene and an allyl cation would be clear-cut examples of single minimum systems. For many other situations it is not so clear whether resonance or equilibrium arrows should be used to indicate the relationship between the two structures.
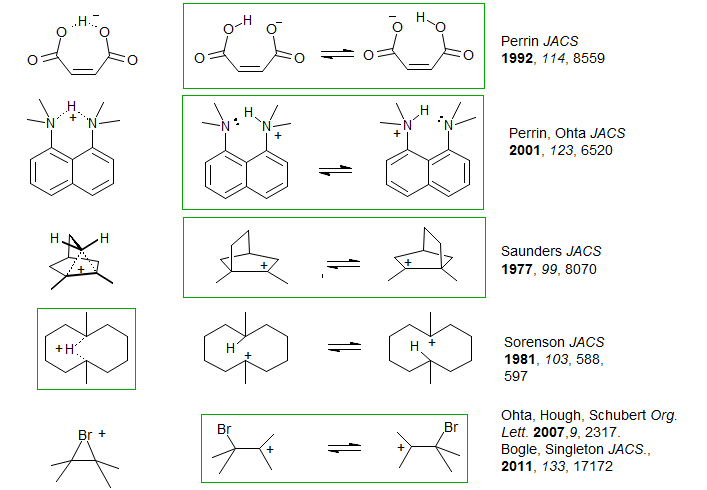
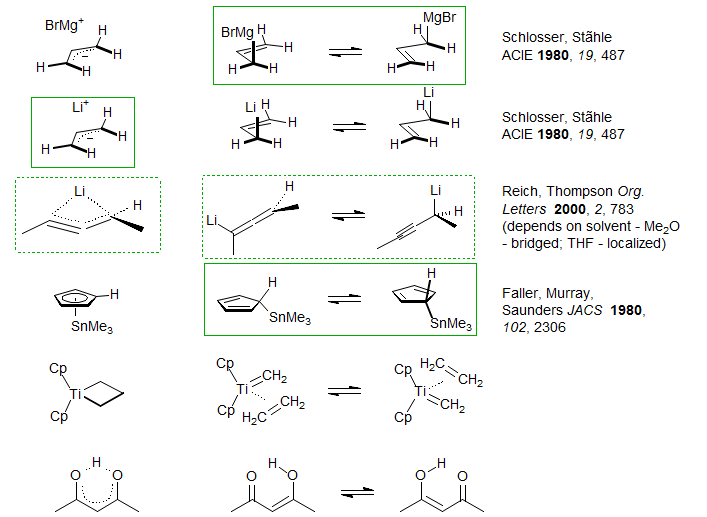
Below are some compounds where this question arises (and the structure favored by the isotope perturbation technique):
If the barrier to interconversion (Ea) is >5 kcal/mol then it is possible to "freeze out" the individual structures in a low temperature (< -140 °C) DNMR experiment, thereby proving that the structure is equilibrating. On the other hand, if the barrier Ea is less than this, then only qualitative arguments using chemical shifts or coupling constants can be made to distinguish the two possibilities since only averaged values will be seen (or a "faster" spectroscopic technique like IR or UV must be applied). The Saunder's equilibrium isotope perturbation technique can often be used to provide an unambiguous answer in such situations ("Studies based on deuterium isotope effect on 13C chemical shifts," T. Dziembowska, P.E. Hansen, Z. Rozwadowski Progr. NMR Spectr. 2004, 45, 1–29).
The effect works if there is an isotopic substitution of an otherwise symmetrical system that stabilizes one of the equilibrating structures versus the other one. Deuterium substitution and observation of 13C NMR signals is by far the most commonly used perturbation, but other isotopes (e.g. 16O/18O, 12C/13C) can also give measurable effects. The perturbation of the equilibrium constant can be due to the greater bond strength of C-D vs C-H (rehydridization, as in allyl organometallics), changes in hyperconjugative effects of C-D vs C-H bonds (as in several carbonium ion systems), or changes in basicity (as in hydrogen bonding systems, where, for example, deuterated amines are more basic than undeuterated). Small equilibrium isotope perturbations are not always easy to distinguished from intrinsic shifts, since the isotopic substitution can itself desymmetrize the structure which is otherwise symmetric, leading to unusually large shifts (Bogle, X. S.; Singleton, D. A. J. Am. Chem. Soc., 2011, 133, 17172–17175)
Carbonium Ions. Consider the carbonium ion system below, one of the earliest and most dramatic examples of the Saunders isotope perturbation effect (Saunders, Telkowski, Kates JACS 1977, 99, 8070). The structure has been desymmetrized by replacing one of the CH3 groups by a CD3. If the structure were delocalized (bridged carbonium ion), then only the normal (intrinsic) isotope shift would be seen. C2 would be upfield of C1 by a fraction of a ppm (perhaps 0.1 ppm per D). On the other hand, if the carbonium ion were an equilibrating structure, then a small deviation from Keq = 1.0 results in a much larger isotope shift - in the calculation below we arbitrarily set Keq = 0.8 (a realistic number) If we assume that the carbonium ion δ is 330 ppm and the quaternary carbon δ is 30 ppm, we calculate the difference between the two carbons (C1 and C2) at 30 ppm.
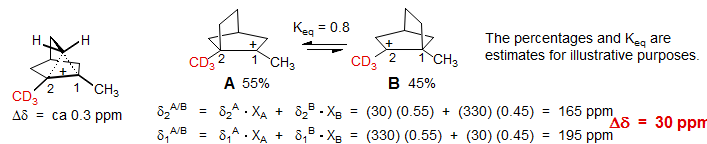
The deviation from Keq = 1.0 arises as follows. Because the C-H bond is weaker than the C-D bond, there will be stronger hyperconjugation between the methyl C-H bonds and the adjacent carbonium ion in A than the C-D bonds and the cationic center in B, i.e., A will be favored.
The 13C NMR spectrum of the dimethylnorbornyl cation is shown below, A Δδ of 35.2 ppm is seen between the CCH3 and CCD3 carbons (C1 and C2). This conclusively rules out a symmetrical structure.
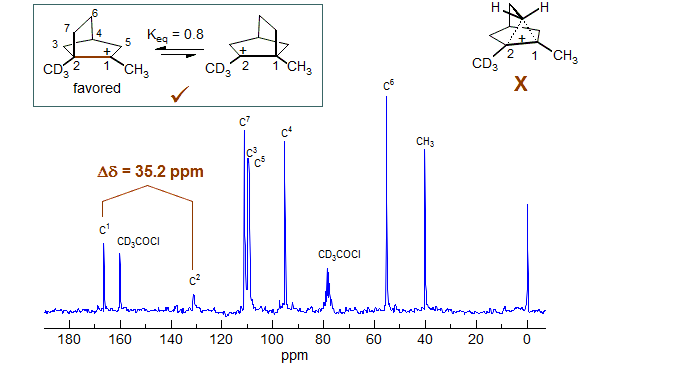
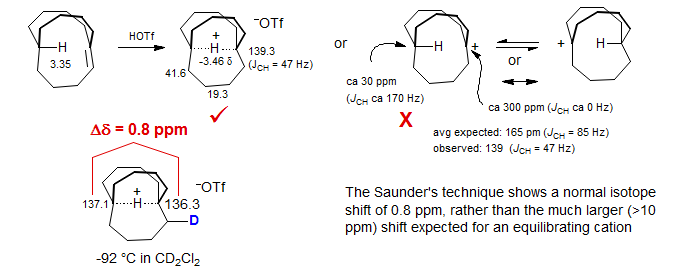
Contrast the above shift with the behavior of the bicyclic hydrogen-bridged delocalized carbonium ion structure shown below (McMurry, Lectka, Hodge J. Am. Chem. Soc. 1989, 111, 8867). Although the bridged nature is quite evident from the chemical shifts, which are far from those expected for an equilibrating structure (expect 165 ppm for equilibratiing structures, observe 139 ppm), as well as the C-H coupling constant (expect 85 Hz for equilibratiing structures, observe 47 Hz), the clearest indication is given by the Saunders isotope perturbation test. The difference between the two bridgehead carbon 13C shifts in the monodeuterated compound shown is 0.8 ppm, a shift in excess of 10 ppm would be expected for an equilibrating structure.
The 13C NMR spectrum of a 1:4 mixture of the H and D substituted cyclohexenyl cations shown below illustrates another non-equilibrating system. The chemical shift difference between the b and c carbons is 29.3 Hz (0.43 ppm). The difference between the a and c carbons is 11.4 Hz (0.17 ppm) (Saunders, Kates JACS 1977, 99, 8071). These are clearly intrinisic shifts.
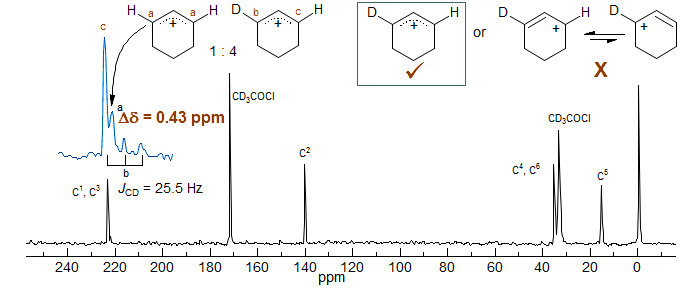
The cyclohexenyl system also shows another interesting effect - the signal c is downfield of the signal a, in contrast to the vast majority of 13C deuterium isotope shifts, which are upfield. This was termed "isotopic perturbation of resonance," where the charge distribution was altered by the presence of H vs D at the other allyl carbon.
Hydrogen Bonding. An example of a symmetric hydrogen bonding situation where both intrinsic and perturbation shifts can be easily distinguished, and which demonstrates an unsymmetrical H-bond even in a geometrically favorable symmetrical structure is given by 1,8-bisdimethylaminonaphthalene (Proton Sponge) (Perrin, Ohta JACS 2001, 123, 6520). The 13C NMR spectrum (DMSO-d6) of the 1,8-carbons of a mixture of compounds with 4 to 0 CH3 and 0 to 4 CD3 groups is shown below.
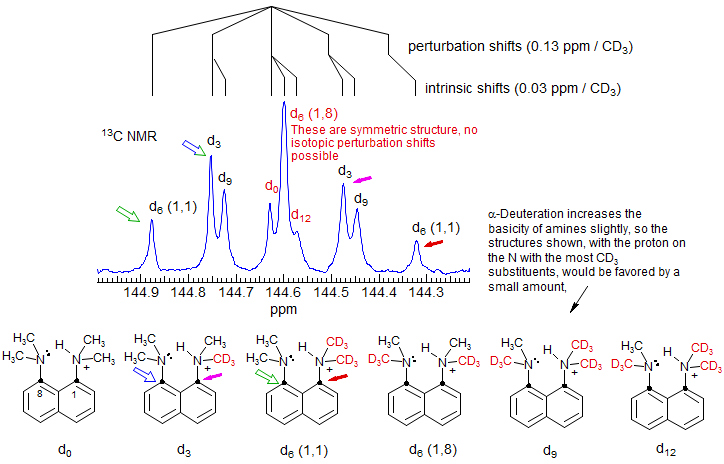
The intrinsic isotope shifts can be identified from the positions of the d0, d6 (1,8) and d12 signals - these are all symmetrically substituted compounds, and so can show no perturbation shifts, only intrinsic ones (in this case 0.03 ppm per CD3). Thus the upfield displacements of the d6 (1,8) and d12 signals are the 3-bond isotope shifts (actually, the sum of the 3-bond and 5-bond shifts, but the 5-bond is expected to be small). The larger displacements of the two d3 signals, and still larger d6 (1,1) signals are then principally the isotope perturbation shifts (0.13 ppm per CD3). The partition into intrinsic and perturbation shifts is illustrated above the spectrum. The upfield peaks correspond to the carbon bonded to the deuterium labeled NMe2 group. This shift is in the expected direction, since α-deuteration increases the basicity of amines (the major isomer for d3, d6 and d9 is expected to be the one shown above), and the carbon bonded to the NHMe2 group is expected to be upfield of the one bonded to the NMe2 group. The observation of significant perturbation shifts show that the hydrogen bond in this compound is unsymmetrical.
Carbanions. The isotope perturbation method can also be applied to situations where the two species in equilibrium are structurally dissimilar, provided both are present in significant concentrations at equilibrium. A case of this type is illustrated by the allenyllithium-propargyllithium system below. The central carbon of allenyllithium reagents is near δ 180 (1, 2), those of propargyllithum near δ 90 (3, 4). Some have intermediate chemical shifts, such as the phenyl-substituted compound 5, with δ 120-140, depending on conditions. So the question is - are such compounds bridged species, or are they equilibrating A and P isomers? For 5, the answer is: it depends on solvent. In THF-Et2O a significant isotope shift is seen for the central carbon in mixtures of H and D-labelled compounds, hence equilibrating structures, whereas in Me2O no splitting can be detected, indicating a single bridged structure . The slightly stronger donor solvent THF favors the equilibrating structure since this allows additional solvation - there is more room for solvent around a mono-coordinated lithium than around a (bridged) dicoordinated one (Reich, Thompson, Org. Letters 2000, 2, 783-786).
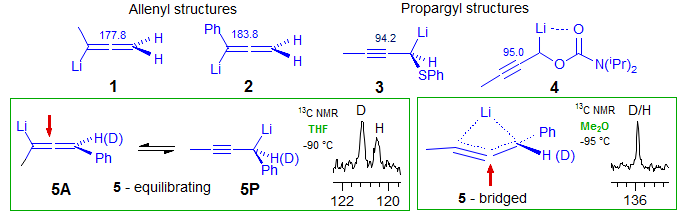
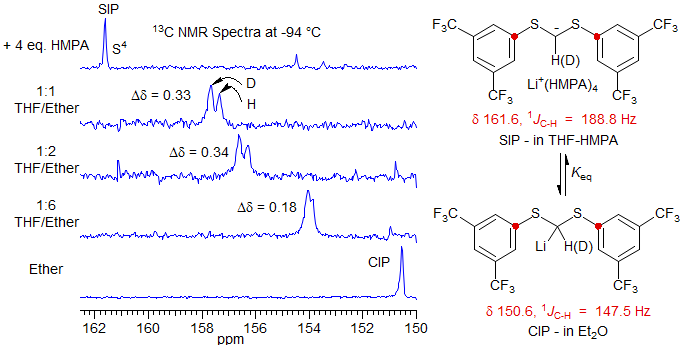
A second example in the carbanion area is provided by a system in which a contact ion pair (CIP) and solvent-separated ion pair (SIP) are shown to be in equilibrium. Since the hybridization at the carbanion carbon changes significantly between the deuterated and undeuterated structures (as shown by the larger JCH in the SIP), there should be a difference in the Keq of the H and D compounds when both are present. Indeed, only a single peak for the C-S carbon is seen for a mixture of H and D compounds in the pure CIP (δ 150.5) and SIP (δ 161.6), indicating that the intrinsic shifts over two bonds are too small to detect. However, at intermediate solvent polarities there are intermediate chemical shifts, indicating a mixture of the two species, and a significant Saunders isotope perturbation shift is observed. The deuterated isotopomer is shifted to lower field because the sp2 carbanion has a stronger C-H/D bond hence more populated) than the lithium reagent (Reich, Sikorski, Thompson, Sanders, Jones, Org. Lett. 2006, 8, 4003-4006). The observed shifts cannot be intrinsic, since these would be in the opposite direction.