8-TECH-7 Lanthanide Induced Shifts (LIS)
β-Diketone complexes of some of the lanthanides have interesting and useful properties as NMR shift reagents. Lewis acid complexation of the lanthanide atom with basic sites on molecules results in substantial chemical shift effects consistent with the presence of large shielding and deshielding cones around the lanthanide atom. These chemical shift effects are the result of unpaired electrons in the f shell of the lanthanide. The lanthanides are especially effective because there is relatively little delocalization of the unpaired f electrons onto the substrate (Fermi contact interactions), and so the principal effect is usually the anisotropy of the metal. An optimum combination of minimum line broadening by direct Fermi contact interaction with unpaired spins, and maximum downfield and upfield dipolar shifts is provided by Eu and Pr tris-β-diketone complexes. The first widely used shift reagent was Eu(dpm)3 (tris(2,2,6,6-tetramethylhepta-3,5-dionato)europium(III)) (Hinckley, J. Amer. Chem. Soc. 1969, 91, 5160). The fluorinated analog Eu(fod)3 (tris(7,7,-dimethyl-1,1,2,2,2,3,3-heptafluoroocta-7,7-dimethyl-4,6-dionato)europium(III) (Rondeau, Sievers, J. Am. Chem. Soc. 1971, 93, 1522) has better solubility and is a stronger Lewis acid.
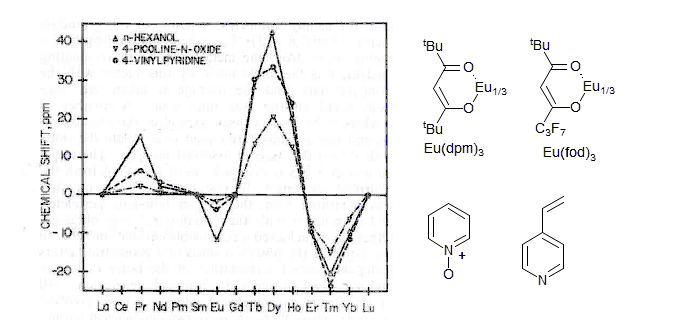
Observed isotropic shifts for the most shifted resonance of 1-hexanol (H-1), 4-picoline-N-oxide (H-2) and 4-vinylpyridine (H-2), in the presence of Ln(dpm)3 at 30 °C in CDCl3.
The choice of Europium as the main shift reagent and Praesidinium as the alternate reagent of choice was dictated by their properties. Eu shows shifts in the downfield direction, thus usually accentuating the existing shift differences in 1H NMR spectra. Pr shifts signals mostly upfield, often initially making the spectra more complicated. The shifts for some of the later lanthanides (Dy and Tm) are much larger, but there are also severe line-broadening effects (with the 2-proton of picoline - 200 Hz for Dy and 65 Hz for Tm, versus ca 5 Hz for Eu and Pr) (Horrocks, W. D, Sipe, J. P. J. Am. Chem. Soc. 1971, 93, 6800).
The chemical shift effects of dipolar interactions are reasonably predicted by the usual deshielding or shielding cone, as for anisotropic effects of various functional groups.
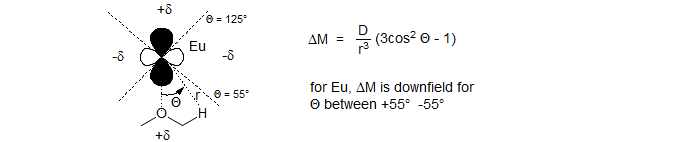
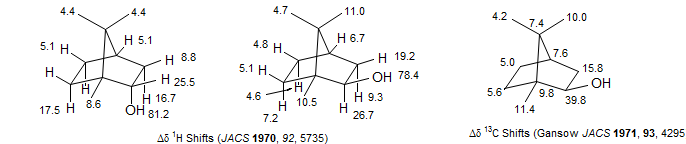
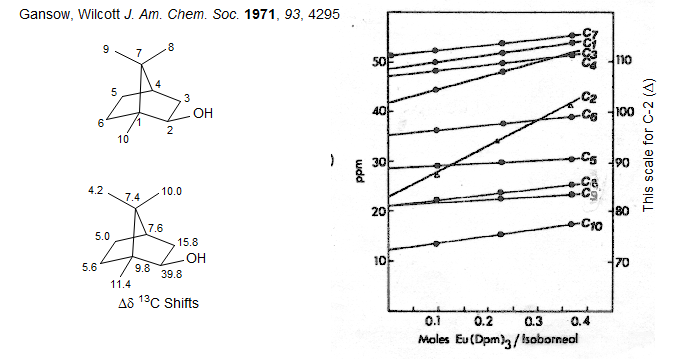
Evidence that the shift effects are primarily due to the magnetic anisotropy of the metal, and not by direct contact interactions with the unpaired spins is provided by the great similarity (in ppm) of the 1H and 13C shifts, as in the example of isoborneol below, with Eu(dpm)3:
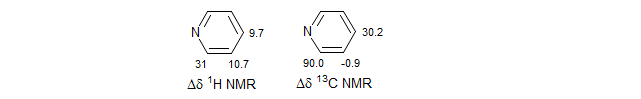
Even the optimized reagents like Eu(dpm)3 show evidence of contact interactions in the 13C shifts of strongly complexing substrates like amines, pyridines and quinolines. Contact effects are undesirable for structure elucidation because they cause line broadening and, in contrast to anisotropy effects, the chemical shifts are not predictable with simple geometric arguments. In fact, they will often alternate signs down a chain of atoms (note the shift changes in the 13C NMR of pyridine). Since the observed shifts are the sum of the contact and dipolar effects, a major contribution from contact effects can interfere with the relatively predictable distance vs shifts effects that make interpretation of the data possible. Contact interactions are less of a problem with 1H NMR spectra.
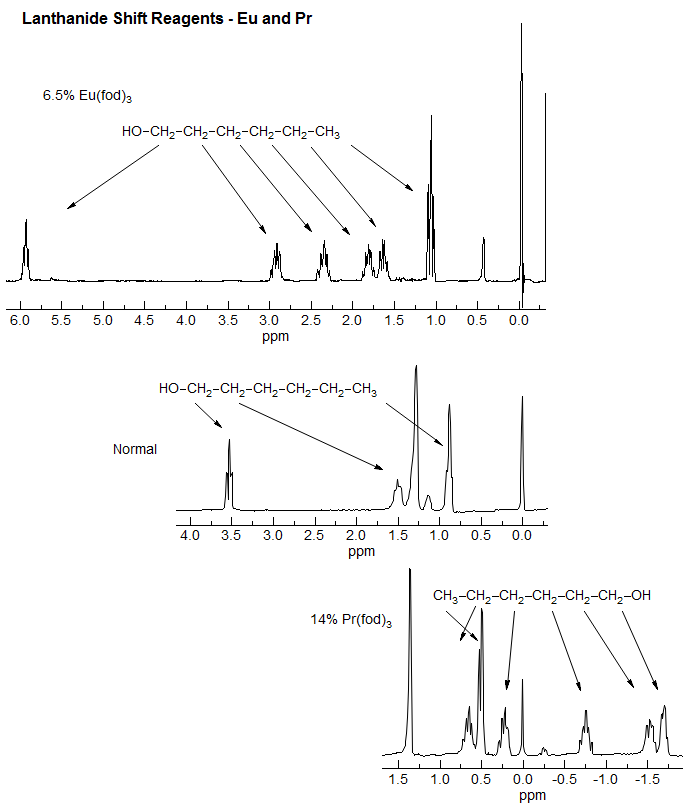
Eu reagents cause mainly downfield shifts, Pr reagents cause upfield shifts. The Eu reagents are much more frequently used, because the shift effects enhance the normal chemical shift differences between protons, whereas Pr reagents initially diminish them. That is, protons near functional groups tend to be downfield of the others, and the Eu shift reagents continue to move them further downfield.
The LIS reagents work only on molecules with Lewis basic sites. Sequence of complexing strength varies with the nature of the substrate, but is approximately: NH2 > OH > R2O > R2C=O > CO2R ≈ R2S > R-CN.
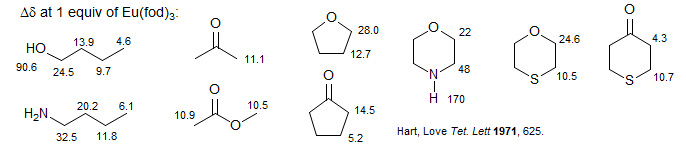
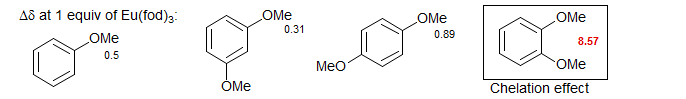
Lanthanide complexes can be 7, 8 or even 9 coordinate. This means that 1:1 and 2:1 substrate to reagent complexes can be formed, and indications of such complexes have been found, for example in nonlinear plots of chemical shift vs mole fraction of shift reagent. The high coordination numbers of the lanthanide complexes also allow chelation to occur when a substrate has more than one suitably situated basic site, as in the case of ortho-dimethoxybenzene below.
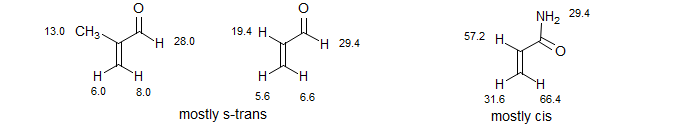
Shift reagents are not used just to simplify spectra. They are especially valuable for distinguishing geometric isomers, such as cis-trans isomers of double bonds. They have sometimes been used for conformational analysis, but this use is constrained by the likelihood that complexation to the lanthanide could cause changes in the conformation of the molecule. In the examples below, the relatively small Δδ values for the beta-hydrogens and large Δδ values of the alpha-substituents of methacrolein and acrolein point to a predominance of the s-trans conformation, whereas the Δδ values for acrylamide suggest an s-cis conformation. However, these conclusions are strictly valid only for the europium complexes, not for the free compounds.
At normal temperatures the complexation and decomplexation rates of typical lanthanide shift reagent-substrate complexes are fast on the NMR time scale, so that averaged signals are obtained. Thus addition of 0.1 molar equivalent of a shift reagent will typically cause about 10% of the chemical shift effect of adding one molar equivalent, since the observed shifts correspond to a weighted average of the 10% strongly shifted molecules with the 90% of unaffected ones.
Chiral Shift Reagents. One of the most useful applications of lanthanide shift reagents is in the determination of optical purity by the use of chiral ligands on the lanthanide. Some of the more effective reagents developed are Eu(facam)3 (tris(3-trifluoroacetyl-d-camphorato)europium(III) and Eu(hfbc)3 (tris(3-heptafluorobutyryl-d-camphorato)europium(III) (H. L. Goering J. Am. Chem. Soc. 1974, 96, 1493). Often sufficient separation between the R and S enantiomers can be obtained so that the enantiomeric purity can be determined directly by NMR integration.
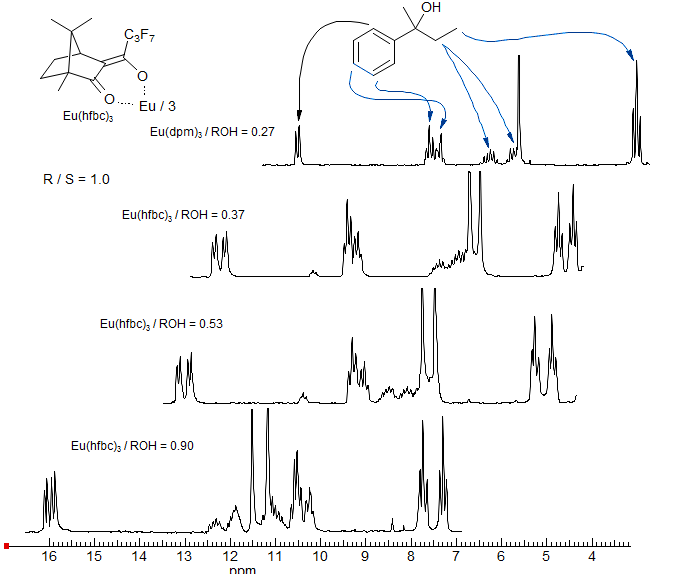
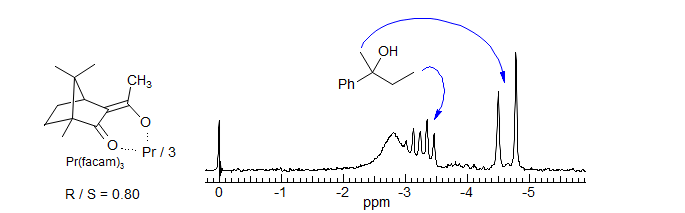
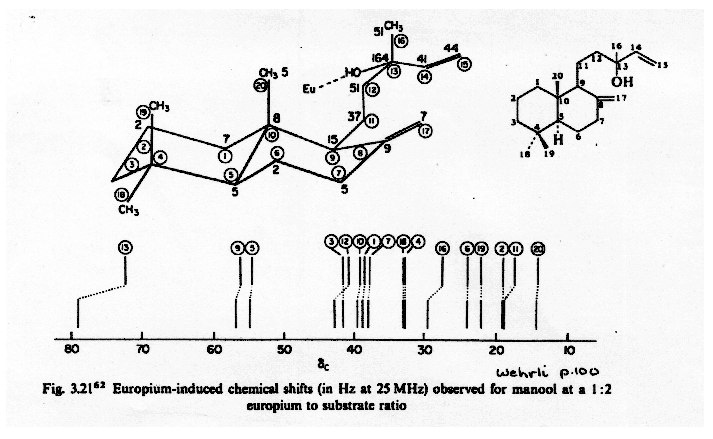
Lanthanide Shift Reagents in 13C NMR Spectroscopy. Since the effect of shift reagents is due to the magnetic anisotropy of the lanthanide metal, the Δδ values for a 13C nucleus are the same in ppm as for a proton in the same location. This means that 13C NMR spectra change much less dramatically than do proton spectra, since the chemical shift range is much larger. Spectral simplification is typically not very dramatic, but shift reagents have been useful for assignment of carbon-13 NMR signals in complex natural products (e.g. Wenkert HCA 1975, 58, 1567).
Using Lanthanide Shift Reagents. Here are some considerations in the practical use of LIS reagents. The solvent must be non-complexing and dry: CCl4, CDCl3 and CD2Cl2 are excellent. Remember that CDCl3 contains a few percent of ethanol-d6 as stabilizer; this must be removed by filtration through a short plug of alumina or silica gel. CDCl3 purified in this way cannot be stored in air for extended periods (days) since it will rapidly autoxidize. The samples must be dry (water complexes strongly to the shift reagents, and causes hydrolysis), and the solution should be filtered to remove paramagnetic particles.
The usual procedure is to prepare a solution of the shift reagent in CDCl3, filter it to remove paramagnetic impurities, and then add small increments of this solution to the solution of the substrate, taking NMR spectra after each addition. In this way it is possible to keep track of the individual signals of the substrate, and determine the Δδ values for all protons of interest.